ABSTRACT
Objective: The sweat test (ST) measures chloride levels in sweat and is considered the gold standard for the diagnosis of cystic fibrosis (CF). However, the reliability of a ST depends on their being performed by experienced technicians and in accordance with strict guidelines. Our aim was to evaluate how sweat stimulation, sweat collection, and chloride measurement are performed at 14 centers (9 public centers and 5 private centers) that routinely perform STs in the state of São Paulo, which has the highest frequency of CF in Brazil. Methods: This was a cross-sectional cohort study, using a standardized questionnaire administered in loco to the staff responsible for conducting STs. Results: No uniformity regarding the procedures was found among the centers. Most centers were noncompliant with the international guidelines, especially regarding the collection of sweat (the samples were insufficient in 10-50% of the subjects tested); availability of stimulation equipment (which was limited at 2 centers); modernity and certification of stimulation equipment (most of the equipment having been used for 3-23 years); and written protocols (which were lacking at 12 centers). Knowledge of ST guidelines was evaluated at only 1 center. Conclusions: Our results show that STs largely deviate from internationally accepted guidelines at the participating centers. Therefore, there is an urgent need for standardization of STs, training of qualified personnel, and acquisition/certification of suitable equipment. These are essential conditions for a reliable diagnosis of CF, especially with the increasing demand due to newborn screening nationwide, and for the assessment of a possible clinical benefit from the use of modulator drugs.
Keywords:
Cystic fibrosis/diagnosis; Cystic fibrosis/prevention & control; Sweat.
RESUMO
Objetivo: O teste do suor (TS) mede os níveis de cloro no suor e é considerado o padrão ouro para o diagnóstico da fibrose cística (FC). Contudo, a confiabilidade do TS depende de sua realização por técnicos experientes e segundo diretrizes rígidas. Nosso objetivo foi avaliar como são realizadas a estimulação e coleta do suor e a dosagem de cloro em 14 centros (9 públicos e 5 privados) que realizam TS rotineiramente no estado de São Paulo, que possui a maior frequência de FC do Brasil. Métodos: Estudo de coorte transversal utilizando um questionário padronizado aplicado in loco ao pessoal responsável pela realização dos TS. Resultados: Não houve uniformidade entre os centros quanto aos procedimentos. A maioria dos centros não era aderente às diretrizes internacionais, especialmente quanto à coleta do suor (amostras insuficientes em 10-50% dos indivíduos testados), disponibilidade de equipamentos de estimulação (limitada em 2 centros), modernidade e certificação dos mesmos (a maioria utilizada há 3-23 anos) e protocolos escritos (ausentes em 12 centros). Avaliou-se o conhecimento sobre diretrizes para TS em apenas 1 centro. Conclusões: Nossos resultados mostram que, nos centros participantes, os TS estão muito distantes das diretrizes internacionalmente aceitas. Portanto, há necessidade urgente de padronização dos TS, de treinamento de pessoal qualificado e de aquisição/certificação de equipamentos adequados. Essas são condições essenciais para um diagnóstico confiável de FC, especialmente com a crescente demanda resultante da triagem neonatal em todo o país, e para a avaliação do possível benefício clínico do uso de moduladores.
Palavras-chave:
Fibrose cística/diagnóstico; Fibrose cística/prevenção & controle; Suor.
INTRODUCTIONThe early observations of salty sweat in cystic fibrosis (CF) led to the development of the "still" gold standard test for CF diagnosis, consisting in the measurement of sweat chloride (Cl−) and sodium (Na+) concentrations. For most of the patients with CF, at least those with classical CF, this assay will reveal elevated levels of both electrolytes, con-firming a diagnosis of CF by this relatively straightforward sweat test (ST).
As in many other countries,(1) the implementation of newborn screening (NBS) for CF in Brazil challenged the diag-nosis paradigm by leading to the routine diagnosis of various asymptomatic children. In 2001, NBS was initiated in some states in Brazil, the Brazilian Unified Health Care System providing a nationwide coverage in 2014 (Appendix 1; available in the online version of the JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48). Although the incidence of CF ranges from 1:2,500 to 1:6,000 live births in Europe and North America,(2) the esti-mated incidence is 1:10,000 live births in Brazil.(3) Based on these data, it is estimated that 60 new CF cases occur per year in the state of São Paulo (SP). NBS for CF caused an increase in the survival of these patients since it ena-bled the early diagnosis of CF and allowed the adoption of nutritional and therapeutic approaches before the advent of clinical manifestations and complications of the disease,(4-6) being economically justifiable for the public health care initiative.(7) In the first month of life of an individual, NBS is carried out by two determinations of the level of immunoreactive trypsinogen. However, the follow-up of the patients with positive results in NBS requires the confir-mation of a diagnosis of CF. This is achieved by ST values ≥ 60 mEq/L in two different samples and/or the identifica-tion of two mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.(8)
Although the ST remains the most sensitive indicator of CF, in order to be considered as the "gold standard", it should be performed using the Gibson and Cooke (GC) technique, also called the quantitative pilocarpine iontopho-resis ST.(9)
For the GC method to be reliable, it needs to be performed in laboratories with experienced and skilled technicians according to strict guidelines,(10) requiring that sweating be stimulated by pilocarpine iontophoresis; sweat is collect-ed on a filter paper or gauze pad, weighed, eluted, and analyzed for Na+ and Cl− using a variety of validated methods described below. The Cystic Fibrosis Foundation summarized its guidelines in 23 topics in order to ensure the appro-priate quality of STs.(11,12) The topics are based on the classical GC method of pilocarpine stimulation,(9) use of filter paper or Macroduct® Sweat Collection System (MSCS; EliTechGroup, Paris, France) for sweat collection, and determi-nation of Cl− by manual titration or by a coulometric quantitative test.(9,10,12-15) The qualitative method is not accepted for confirming a definitive diagnosis of CF.(10,13,16)
Indeed, the ST is complex, and its accuracy is related to the competence and commitment of the professional who carries out its various steps.(9,10) For this reason, various countries organized standardized protocols for STs. The first country to publish a consensus standardization and external quality control for STs was the USA in 1994, followed by the United Kingdom in 2000.(17-20) Since then, numerous guidelines have been published, enforcing specific rules to be adopted while carrying out STs, as well as demanding accreditation and periodic monitoring of the laboratories by official regulatory agencies.(13)
Nevertheless, even in countries where the standardization of STs is well established, there are details in the per-formance and interpretation of the tests that are often omitted and overlooked from center to center.(12,21) Moreover, the ST has also become a major outcome measure in clinical trials, namely those involving CFTR modulators that rescue the function of the dysfunctional mutant protein.(22,23) Therefore, it has become increasingly relevant that the performance and the standard procedures of STs should be reviewed at present times.
Taking into account the socioeconomic status and the miscegenation of the Brazilian population, the importance of STs and other methods for the diagnosis of CF is even more relevant.(24-28) In Brazil, to date, there is no critical and comparative analysis regarding the performance and the interpretation of STs. We selected the state of SP because it is the most populous in Brazil, with approximately 45 million inhabitants in 2016 (Appendix 2; available in the online version of the JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48). (29)
The objective of the present study was to evaluate how STs are performed and interpreted at the centers that agreed to participate in the study and that routinely perform these tests in various cities in the state of SP, comparing their routine with those specified in international guidelines. These centers, altogether, carry out approximately 4,500 tests per year.
METHODS
This was an observational, descriptive, cross-sectional cohort study. A total of 18 centers that routinely perform STs in the state of SP were invited to participate in the study in 2013. The study was approved by the Research Ethics Committee of the Universidade Estadual de Campinas (State University of Campinas; Protocol no. 86624/2012) and was carried out in accordance with the Declaration of Helsinki. All participants gave written informed consent.
A questionnaire was developed, consisting of 54 questions that comprised all of the steps for performing STs: sweat stimulation; collection of sweat; and determination of the level of Cl−(12,13) (Appendix 3; available in the online version of the JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48). . The questionnaire was given to the staff responsible for conducting the STs at all of the centers included in the study. In our study, two researchers performed the interviews simultaneously. The data were compiled in Excel spreadsheets, and the results were pre-sented in tables and figures.
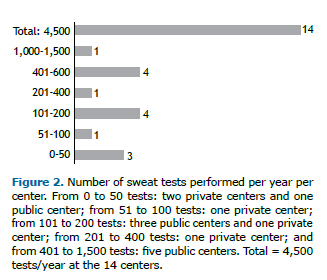
RESULTSA total of 18 centers were identified as performing STs as part of their routine (9 were private and 9 were public). Of the 18 centers, 14 agreed to participate in the study, 5 (35.7%) being private institutions and 9 (64.3%) being public health care centers (Figure 1). At the moment of the visit, 4 of the centers were not performing STs by lack of supplies. The 14 centers included in the study perform approximately 4,500 STs/year, 4,000 of those being carried out at public centers. The number of STs/year per center is shown in Figure 2. Among the 14 centers, the length of experience in performing STs ranged from 1 to > 20 years (Appendix 4; available in the online version of the JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48).

We interviewed the professionals involved in the performance of the three stages of STs at the participating centers. Regarding their occupation, one was a physician, seven were biomedical technicians, two were biologists, four were nurses, and seven were nursing technicians. Among the 14 centers, 11 professionals were trained by colleagues from the same center (internal training), 2 were trained at another center (external training), and 1 had both internal and external training. Only 1 of the centers was aware of the ST guidelines and had a printed version of the standard operating procedure manual.
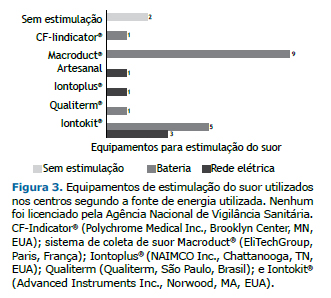
Among the 14 centers, 2 had no equipment for sweat stimulation. Some centers used more than one device, the equipment used being nine MSCS; one CF-Indicator® (Polychrome Medical Inc., Brooklyn Center, MN, USA); five Iontokit® (Advanced Instruments Inc., Norwood, MA, USA); and one produced by Qualiterm (Qualiterm, São Paulo, Brazil). The five devices connected to a power grid were as follows: three Iontokit® (Advanced Instruments Inc.), one Iontoplus® (NAIMCO Inc., Chattanooga, TN, USA); and one handmade craft. These devices were older, with a mean usage time of 15 years (Figure 3).
There were 21 pieces of stimulation equipment at 12 of the centers altogether. None of the pieces of equipment had an official registration for the clinical diagnosis of CF in the country. Among these, there were 8 MSCS, 7 of which not being in operation due to lack of spare parts or supplies, or because they broke down less than a year ago. Among the 12 centers that carried out sweat stimulation, 8 used the forearm/arm for electrode placement, whereas the other 4 used other sites (Figure 4A). Sweat stimulation was achieved by one or more of the following techniques: use of a blanket, at 6 centers; use of a coat, at 6; running or walking outdoors, at 4; skin lock with plastic wrap, at 2; use of a bandage, at 1; and Parafilm wrapping, at 1. Sweat was collected from the patient with the help of a disposable spoon and placed into a sterile tube for the determination of the level of Cl− at 1 center. At 2 private centers, sweat was induced without stimulation by iontophoresis devices (Appendix 5; available in the online version of the JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48).
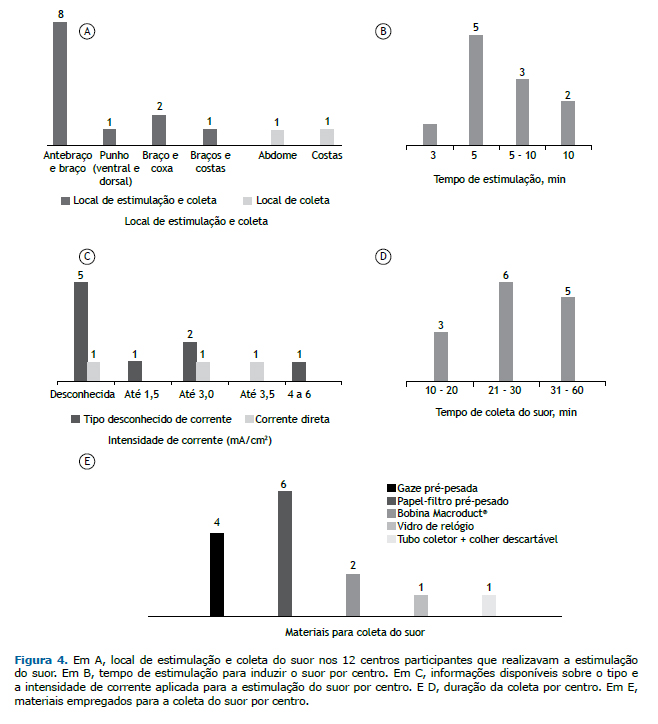
The electrical stimulation time to induce sweat ranged from 3 to 10 min (Figure 4B). The type of current used was known at only 3 of the 14 centers (direct current) and unknown at 9, 5 of which also failed to report the intensity of the current used. The intensity of the current was known at 5 centers (Figure 4C). At 4 centers (28.6%), it was report-ed that the electrical stimulation procedure had caused skin burn in some patients; however, 5 centers (35.7%) re-ported that it had never occurred with any of their patients, whereas 3 centers (21.4%) were unable to report that information since the interviewees had been recently working at those centers. Finally, 2 centers (14.3%) did not use electrical stimulation.
The duration of sweat collection ranged from 10 to 60 min (Figure 4D). The materials employed for sweat collection at the 14 centers are described in Figure 4E. It is of note that 2 centers were using unusual materials/techniques: a watch glass was placed over the site of pilocarpine stimulation, then fixed with tape, and the sweat droplets were collected using a sterile pipette or a disposable spoon into a sterile tube at 1 of the centers, whereas the other center did not have appropriate precision scales for weighing the collected material. In addition, 2 centers did not perform sweat stimulation using any kind of equipment and, therefore, were disregarded regarding this issue.
The Cl− level was determined by using the manual titration technique and the quantitative coulometric test (chlo-ridometer), at 6 centers each. Among the latter, 1 center was not in operation. The need to repeat a ST was due to insufficient sweating and unknown causes at 10 and 4 of the centers, respectively. The rate of repeat STs was 5%, at 1 center; 10-20%, at 7; 30%, at 1; and > 50%, at 1.
The number of people who collected sweat and conducted the STs is summarized in Figure 5A. The same profes-sional was responsible for the collection and the performance of the ST at 6 centers. The minimum acceptable amount of sweat in order to conduct a reliable ST considerably varied among the centers and is summarized in Figure 5B. At 11 centers that used filter paper or gauze to collect sweat, the amount of sweat considerably ranged from 50 to 100 mg, and 1 center had no knowledge of the acceptable value (Figure 5B). Regarding the 3 centers using the MSCS, the acceptable volume of sweat was 15 µL, 20 µL, and no standard. The center that used the watch glass reported that the required minimum volume was 20 µL. Most of the professionals at the centers were unaware of the correct number of tests with positive results (Cl− ≥ 60 mEq/L) for a definitive diagnosis of CF (Figure 5C). Figure 6 shows how compliant with the Cystic Fibrosis Foundation guidelines(13) each of the 14 centers was.

 DISCUSSION
DISCUSSIONIn Brazil, the diversity of expression of the disease is conditioned by miscegenation, which highly increases the genetic diversity expressed in a variability of mutations in the CFTR gene in our population, thus rendering the genet-ic diagnosis difficult and costly. The monitoring of CF patients in Brazil is performed at referral centers, most of which are public and linked to the Brazilian Unified National Health Care System with public financial support and generally associated with universities. The state of SP has the second per capita income in the country and has the largest number of referral centers for CF (n = 7), all of which being public health care centers (Appendix 6 ; available in the online version of the JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48).
The present study on the performance of STs at 14 centers, which perform 4,500 tests/year altogether, revealed that there is no uniformity in the ST procedures and that there are serious difficulties in its performance and signifi-cantly inadequate conditions, which largely deviate it from internationally accepted guidelines.
Similar studies, however, had been previously performed regarding the quality of STs at various centers in several other countries, also presenting significant diversity and inconsistent results.(12,30,31) The diagnostic confirmation of CF enables health care centers to provide better care and monitoring, which translates into higher life expectancy for the patients. (8) This was also found at our referral center.(32) The standardization of STs is key to a reliable diagnosis of CF.
In particular, the present study has shown that there is no uniformity in the performance of ST in its three stages (stimulation, collection, and quantification) at the participating centers. The main issues and possible alternatives to the three steps can be summarized as follows:
(i) Stimulation: the lack of awareness about the existence of adequate equipment for sweat stimulation and its us-age was present at approximately 30% of the participating centers. Alternative methods of stimulation, non-compliant with the international guidelines (e.g. exposure to the sun with a blanket, exposure to the sun inside a car, usage of noncertified sweat induction equipment, and lack of knowledge of stimulation techniques), were in practice at 4 of the centers and might even impair the health of patients (e.g. skin burning, dehydration, or even death). In addition, those 4 centers also collected inadequate sweat samples for determining the level of Cl−. Every center should rely on the iontophoresis stimulation technique with pilocarpine, use certified stimula-tion equipment, and carry out its regular maintenance and calibration by the manufacturer or a qualified com-pany in order to ensure the safety of the procedure. Every center should also provide (or seek elsewhere) ade-quate training of the professionals responsible for handling the equipment.
(ii) Collection: most of the participating centers used methods for sweat collection that were in accordance with the established recommendations (use of filter paper, gauze, or MSCS). However, 2 centers used alternative, non-recommended collection methods (use of a spoon and a watch glass), which affect the reliability of the ST. Alt-hough MSCS has been described as a suitable method for the collection of sweat by the US Health Department since 2013, it was not registered in Brazil during the period of study.(13) In September of 2014, the use of MSCS was properly licensed, as well as of the digital chloridometer, which enabled adequate, periodic maintenance of the equipment and uninterrupted acquisition of supplies. This fact will possibly change the present scenario: 7 centers were not using MSCS due to the lack of spare parts/supplies, difficulties in the maintenance of the equipment, or lack of trained professionals capable of using the system (how to perform sweat induction, sweat collection, and Cl− level determination). Another factor to be considered is the high cost of supplies, which are imported, in comparison with the traditional method of stimulation and collection for the GC method, which uses pilocarpine and filter paper or gauze for sweat collection.
(iii) Quantification: The determination of the Cl− level must be quantitative and performed by coulometry, flame photometry, or manual titration. Regardless of the procedure, there were no problems regarding this issue at the participating centers, except that conductivity, which is an unreliable procedure, was used for the diagnosis of CF at 2 centers. At another center, conductivity was used just for screening, which is an appropriate, correct approach.
CF is a progressive disease, which requires that patients be treated at referral centers so as to receive the best health care and adequate treatment. Having a safe, reliable diagnosis is the first step, and it is critical for the guid-ance of patients and their families by the medical staff.
The present study shows the real situation of STs in the state of SP, which may be representative of the overall sit-uation for STs in Brazil. Altogether, our results show that STs largely deviate from internationally accepted guidelines at the participating centers. There is an urgent need for either domestic or imported equipment for sweat stimulation and for determining the level of Cl− in sweat in accordance with international guidelines. Maintenance should be adequate, and spare parts and supplies should be always available so that the results obtained are reliable and appropriate.
From this unique moment, when we celebrate the introduction of NBS for CF in all states in Brazil, we must be pre-pared to overcome the challenges ahead. These challenges can only be overcome if they are first identified and objectively faced by the CF teams. In addition, we need to work together so as to change the current reality; we believe that we can only build a new reality if we first become aware of the limitations and difficulties inherent to each individual center.
The present study provides a warning regarding the need for standardization of STs in Brazil by means of the con-struction and adoption of guidelines for the diagnosis of CF. The issue has been in the discussion agenda of the Bra-zilian CF Study Group.
Directors of CF referral centers should review each current step in the performance of STs, working closely together with the laboratory staff, which will undoubtedly improve the quality of the results so as to provide a reliable CF diagnosis and minimize the possible bias of STs. The professionals who perform STs should be aware of the specificities of the disease and recognize the important role of a correctly performed ST to confirm or exclude a diagnosis of CF. Such training should be provided by the CF referral centers to its professionals, fostering their participation in internal or external training, scientific events, and other discussion forums. The proximity among the different profes-sionals working in the area of CF, as well as the exchange of information among the different referral centers, will allow to increasing the knowledge regarding STs and, therefore, improve the procedures involved in it toward a more reliable diagnosis of CF.
In conclusion, we found no uniformity in the steps carried out in the performance of STs and large deviations from internationally accepted ST guidelines at the various participating centers in the state of SP. Major inadequate condi-tions included insufficient production of sweat, lack of stimulation equipment or clinical chemistry equipment, absence of written protocols, and use of obsolete noncertified measuring equipment. Even though we know that there are various difficulties and barriers to be overcome, we need to move towards meeting the 23 topics of the Cystic Fibrosis Foundation guidelines(13) so that STs are adequately carried out to this end. There is an urgent need for standardiza-tion of STs, training of qualified personnel, suitable equipment, and certification. These are essential conditions for a reliable diagnosis of CF, especially with the increasing demand due to NBS nationwide, and for the assessment of a possible clinical benefit with the use of CFTR modulator drugs.
ACKNOWLEDGEMENTSCystic Fibrosis Collaborative Study Group: Francisco Ubaldo Vieira Junior, Margarida Duarte Amaral, Neiva Dama-ceno, Joaquim Carlos Rodrigues, Giesela Fleischer Ferrari, Vera Lucia Sdepanian, João Batista Salomão Junior, Sônia Mayumi Chiba, Sônia Letícia Silva Lorena, and Maria Inêz Machado Fernandes.
REFERENCES1. Levy H, Farrell PM. New challenges in the diagnosis and management of cystic fibrosis. J Pediatr. 2015;166(6):1337-41. https://doi.org/10.1016/j.jpeds.2015.03.042
2. Cystic Fibrosis Data Network [homepage on the Internet]. Belconnen, Australia: Geoff Sims Consulting Pty Ltd.; c2013 [cited 2016 Feb 2]. Cystic fibrosis data. Available: http://www.cysticfibrosisdata.org/Home.htm
3. Raskin S, Pereira-Ferrari L, Reis FC, Abreu F, Marostica P, Rozov T, et al. Incidence of cystic fibrosis in five different states of Brazil as determined by screening of p.F508del, mutation at the CFTR gene in newborns and patients. J Cyst Fibros. 2008;7(1):15-22. https://doi.org/10.1016/j.jcf.2007.03.006
4. Camargos P, Gomes DL, Alvim CG, Gomes FS, Cajazeiro JM. From lip to lab: salty tasting skin is the main clue that raises clinical suspicion of cystic fibrosis in young infants. Acta Paediatr. 2015;104(5):e210-5. https://doi.org/10.1111/apa.12958
5. Dijk FN, Fitzgerald DA. The impact of newborn screening and earlier intervention on the clinical course of cystic fibrosis. Paediatr Respir Rev. 2012;13(4):220-5. https://doi.org/10.1016/j.prrv.2012.05.003
6. Vernooij-van Langen AM, Gerzon FL, Loeber JG, Dompeling E, Dankert-Roelse JE. Differences in clinical condition and genotype at time of diagnosis of cystic fibrosis by newborn screening or by symptoms. Mol Genet Metab. 2014;113(1-2):100-4. https://doi.org/10.1016/j.ymgme.2014.07.012
7. van der Ploeg CP, van den Akker-van Marle ME, Vernooij-van Langen AM, Elvers LH, Gille JJ, Verkerk PH, et al. Cost-effectiveness of newborn screening for cystic fibrosis determined with real-life data. J Cyst Fibros. 2015;14(2):194-202. https://doi.org/10.1016/j.jcf.2014.08.007
8. Smyth AR, Bell SC, Bojcin S, Bryon M, Duff A, Flume P, et al. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J Cyst Fibros. 2014;13 Suppl 1:S23-42. https://doi.org/10.1016/j.jcf.2014.03.010
9. Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics. 1959;23(3):545-9.
10. Collie JT, Massie RJ, Jones OA, LeGrys VA, Greaves RF. Sixty-five years since the New York heat wave: advances in sweat testing for cystic fibrosis. Pediatr Pulmonol. 2014;49(2):106-17. https://doi.org/10.1002/ppul.22945
11. Cystic Fibrosis Foundation [homepage on the Internet]. Bethesda: the Foundation; [cited 2016 Feb 2]. Available: https://www.cff.org/
12. Cirilli N, Padoan R, Raia V; ICFS Sweat Test Working Group. Audit of sweat testing: a first report from Italian Cystic Fibrosis Centres. J Cyst Fibros. 2008;7(5):415-22. https://doi.org/10.1016/j.jcf.2008.03.005
13. LeGrys VA, Yankaskas JR, Quittell LM, Marshall BC, Mogayzel PJ Jr; Cystic Fibrosis Foundation. Diagnostic sweat testing: the Cystic Fibrosis Foundation guidelines. J Pediatr. 2007;151(1):85-9. https://doi.org/10.1016/j.jpeds.2007.03.002
14. Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153(2):S4-S14. https://doi.org/10.1016/j.jpeds.2008.05.005
15. Castellani C, Southern KW, Brownlee K, Dankert Roelse J, Duff A, Farrell M, et al. European best practice guidelines for cystic fibrosis neonatal screening. J Cyst Fibros. 2009;8(3):153-73 https://doi.org/10.1016/j.jcf.2009.01.004
16. Lezana JL, Vargas MH, Karam-Bechara J, Aldana RS, Furuya ME. Sweat conductivity and chloride titration for cystic fibrosis diagnosis in 3834 subjects. J Cyst Fibros. 2003;2(1):1-7. https://doi.org/10.1016/S1569-1993(02)00146-7
17. US National Committee for Clinical Laboratory Standards. Sweat testing: sample collection and quantitative analysis; approved guideline. 2nd ed. Waynes (PA): US National Committee for Clinical Laboratory Standards; 2000 Jun. Document No: C34-A2.
18. US National Committee for Clinical Laboratory Standards. Sweat testing: sample collection and quantitative analysis; approved guideline. 3rd ed. Waynes (PA): US National Committee for Clinical Laboratory Standards; 2009 Dec. Document No: C34-A3.
19. LeGrys VA. Sweat analysis proficiency testing for cystic fibrosis. Pediatr Pulmonol. 2000;30(6):476-80. https://doi.org/10.1002/1099-0496(200012)30:6<476::AID-PPUL7>3.0.CO;2-O
20. LeGrys VA. Assessment of sweat-testing practices for the diagnosis of cystic fibrosis. Arch Pathol Lab Med. 2001;125(11):1420-4.
21. Baumer JH. Evidence based guidelines for the performance of the sweat test for the investigation of cystic fibrosis in the UK. Arch Dis Child. 2003;88(12):1126-7. https://doi.org/10.1136/adc.88.12.1126
22. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663-72. https://doi.org/10.1056/NEJMoa1105185
23. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220-31. https://doi.org/10.1056/NEJMoa1409547
24. Quinton P, Molyneux L, Ip W, Dupuis A, Avolio J, Tullis E, et al. -adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am J Respir Crit Care Med. 2012;186(8):732-9. https://doi.org/10.1164/rccm.201205-0922OC
25. Gonska T, Ip W, Turner D, Han WS, Rose J, Durie P, et al. Sweat gland bioelectrics differ in cystic fibrosis: a new concept for potential diagnosis and assessment of CFTR function in cystic fibrosis. Thorax. 2009;64(11):932-8. https://doi.org/10.1136/thx.2009.115295
26. Sousa, M, Servidoni MF, Vinagre A, Ramalho AS, Bonadia LC, Felício V, et al. Measurements of CFTR-mediated Cl- secretion in human rectal biopsies constitute a robust biomarker for Cystic Fibrosis diagnosis and prognosis. PLoS One. 2012;7(10):e47708. https://doi.org/10.1371/journal.pone.0047708
27. Gonçalves AC, Marson FA, Mendonça RM, Ribeiro JD, Ribeiro AF, Paschoal IA, et al. Saliva as a potential tool for cystic fibrosis diagnosis. Diagn Pathol. 2013;8:46. https://doi.org/10.1186/1746-1596-8-46
28. Ng RT, Marson FA, Ribeiro JD, Ribeiro AF, Bertuzzo CS, Ribeiro MA, et al. Nasal potential difference in cystic fibrosis considering severe CFTR mutations. Dis Markers. 2015;2015:306825
29. Instituto Brasileiro de Geografia e Estatística [homepage on the Internet]. São Paulo: IBGE; c2016 [cited 2016 Feb 2]. Estados@--São Paulo: Botucatu. [about 3 screens]. Available from: http://www.ibge.gov.br/estadosat/perfil.php?sigla=sp
30. Kirk JM. Inconsistencies in sweat testing in UK laboratories. Arch Dis Child. 2000;82(5):425-7. https://doi.org/10.1136/adc.82.5.425
31. Mackay R, George P, Kirk J. Sweat testing for cystic fibrosis: A review of New Zealand laboratories. J Paediatr Child Health. 2006;42(4):160-4. https://doi.org/10.1111/j.1440-1754.2006.00822.x
32. Marson FA, Hortencio TD, Aguiar KC, Ribeiro JD; CYFIUC Group. Demographic, clinical, and laboratory parameters of cystic fibrosis during the last two decades: a compara-tive analysis. BMC Pulm Med. 2015;15:3. https://doi.org/10.1186/1471-2466-15-3











 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket