ABSTRACT
Objective: The sweat test (ST) measures chloride levels in sweat and is considered the gold standard for the diagnosis of cystic fibrosis (CF). However, the reliability of a ST depends on their being performed by experienced technicians and in accordance with strict guidelines. Our aim was to evaluate how sweat stimulation, sweat collection, and chloride measurement are performed at 14 centers (9 public centers and 5 private centers) that routinely perform STs in the state of São Paulo, which has the highest frequency of CF in Brazil. Methods: This was a cross-sectional cohort study, using a standardized questionnaire administered in loco to the staff responsible for conducting STs. Results: No uniformity regarding the procedures was found among the centers. Most centers were noncompliant with the international guidelines, especially regarding the collection of sweat (the samples were insufficient in 10-50% of the subjects tested); availability of stimulation equipment (which was limited at 2 centers); modernity and certification of stimulation equipment (most of the equipment having been used for 3-23 years); and written protocols (which were lacking at 12 centers). Knowledge of ST guidelines was evaluated at only 1 center. Conclusions: Our results show that STs largely deviate from internationally accepted guidelines at the participating centers. Therefore, there is an urgent need for standardization of STs, training of qualified personnel, and acquisition/certification of suitable equipment. These are essential conditions for a reliable diagnosis of CF, especially with the increasing demand due to newborn screening nationwide, and for the assessment of a possible clinical benefit from the use of modulator drugs.
Keywords:
Cystic fibrosis/diagnosis; Cystic fibrosis/prevention & control; Sweat.
RESUMO
Objetivo: O teste do suor (TS) mede os níveis de cloro no suor e é considerado o padrão ouro para o diagnóstico da fibrose cística (FC). Contudo, a confiabilidade do TS depende de sua realização por técnicos experientes e segundo diretrizes rígidas. Nosso objetivo foi avaliar como são realizadas a estimulação e coleta do suor e a dosagem de cloro em 14 centros (9 públicos e 5 privados) que realizam TS rotineiramente no estado de São Paulo, que possui a maior frequência de FC do Brasil. Métodos: Estudo de coorte transversal utilizando um questionário padronizado aplicado in loco ao pessoal responsável pela realização dos TS. Resultados: Não houve uniformidade entre os centros quanto aos procedimentos. A maioria dos centros não era aderente às diretrizes internacionais, especialmente quanto à coleta do suor (amostras insuficientes em 10-50% dos indivíduos testados), disponibilidade de equipamentos de estimulação (limitada em 2 centros), modernidade e certificação dos mesmos (a maioria utilizada há 3-23 anos) e protocolos escritos (ausentes em 12 centros). Avaliou-se o conhecimento sobre diretrizes para TS em apenas 1 centro. Conclusões: Nossos resultados mostram que, nos centros participantes, os TS estão muito distantes das diretrizes internacionalmente aceitas. Portanto, há necessidade urgente de padronização dos TS, de treinamento de pessoal qualificado e de aquisição/certificação de equipamentos adequados. Essas são condições essenciais para um diagnóstico confiável de FC, especialmente com a crescente demanda resultante da triagem neonatal em todo o país, e para a avaliação do possível benefício clínico do uso de moduladores.
Palavras-chave:
Fibrose cística/diagnóstico; Fibrose cística/prevenção & controle; Suor.
INTRODUÇÃOAs primeiras observações de suor salgado na fibrose cística (FC) levaram ao desenvolvimento do "ainda" teste padrão ouro para o diagnóstico da FC, que consiste na dosagem de cloro (Cl−) e sódio (Na+) no suor. Para a maioria dos pacientes com FC, pelo menos aqueles com FC clássica, esse ensaio irá revelar níveis elevados de ambos os eletrólitos, confirmando o diagnóstico de FC por meio desse teste do suor (TS) relativamente simples.
Assim como em muitos outros países,(1) a implementação da triagem neonatal (TN) no Brasil desafiou o paradigma diagnóstico ao levar ao diagnóstico rotineiro de diversas crianças assintomáticas. Em 2001, foi iniciada a TN em alguns estados do Brasil, com cobertura nacional pelo Sistema Único de Saúde em 2014 (Anexo 1; disponível na versão on-line do JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48). Embora a inci-dência de FC varie de 1:2.500 a 1:6.000 nascidos vivos na Europa e na América do Norte,(2) a incidência estimada no Brasil é de 1:10.000 nascidos vivos. (3) Com base nesses dados, estima-se que ocorram 60 novos casos de FC por ano no estado de São Paulo (SP). A TN para FC causou um aumento na sobrevida desses pacientes, pois possibilitou o diagnóstico precoce da FC e permitiu a adoção de abordagens nutricionais e terapêuticas antes do surgimento das manifestações clínicas e complicações da doença,(4-6) sendo economicamente justificável para a iniciativa de saúde pública.(7) No primeiro mês de vida de um indivíduo, a TN é realizada por meio de duas dosagens de tripsinogênio imunorreativo. Contudo, o acompanhamento dos pacientes com resultados positivos na TN exige a confirmação do diagnóstico de FC. Isso é conseguido por meio de valores ≥ 60 mEq/L no TS em duas amostras diferentes e/ou a identificação de duas mutações no gene cystic fibrosis transmembrane conductance regulator (CFTR).(8)
Embora o TS continue sendo o indicador mais sensível da FC, para que seja considerado o "padrão ouro", ele deve ser realizado segundo a técnica de Gibson & Cooke (GC), também denominada TS por iontoforese quantitativa com pilocarpina.(9)
Para que o método de GC seja confiável, ele precisa ser realizado em laboratórios com técnicos experientes e ha-bilidosos, segundo diretrizes rígidas,(10) exigindo que o suor seja estimulado por iontoforese com pilocarpina; o suor é coletado em papel-filtro ou gaze, pesado, eluído e analisado para Na+ e Cl− por meio de uma variedade de méto-dos validados descritos abaixo. A Cystic Fibrosis Foundation resumiu suas diretrizes em 23 tópicos para garantir a qualidade apropriada dos TS.(11,12) Os tópicos baseiam-se no método clássico de GC de estimulação com pilocarpi-na,(9) no uso de papel-filtro ou do Macroduct® Sweat Collection System (MSCS, sistema de coleta de suor Macroduct®; EliTechGroup, Paris, França) para a coleta do suor e na dosagem de Cl− por titulação manual ou teste quantitativo coulométrico.(9,10,12-15) O método qualitativo não é aceito para confirmação do diagnóstico definitivo de FC.(10,13,16)
De fato, o TS é complexo, e sua acurácia está relacionada à competência e ao comprometimento dos profissionais que realizam suas diversas etapas. (9,10) Por isso, diversos países organizaram protocolos padronizados para os TS. O primeiro país a publicar uma padronização de consenso e controle externo de qualidade para os TS foram os EUA em 1994, seguidos pelo Reino Unido em 2000.(17-20) Desde então, inúmeras diretrizes foram publicadas, com aplica-ção de regras específicas a serem adotadas durante a realização dos TS e também a exigência de acreditação e acompanhamento periódico dos laboratórios por agências reguladoras oficiais.(13)
Não obstante, mesmo em países onde a padronização dos TS já está bem estabelecida, há detalhes da realização e interpretação dos testes que são frequentemente omitidos ou negligenciados de um centro para o outro.(12,21) Além do mais, os TS também se tornaram a principal medida de desfecho em ensaios clínicos, isto é, aqueles envolvendo moduladores de CFTR que resgatam a função da proteína mutante disfuncional. (22,23) Portanto, torna-se cada vez mais relevante que a realização e os procedimentos padrão dos TS sejam revisados na atualidade.
Levando-se em consideração o nível socioeconômico e a miscigenação da população brasileira, a importância dos TS e outros métodos para o diagnóstico da FC é ainda mais relevante.(24-28) No Brasil, até o momento, não há uma análise crítica e comparativa sobre a realização e interpretação dos TS. Selecionamos o estado de SP por ele ser o mais populoso do Brasil, com aproximadamente 45 milhões de habitantes em 2016 (Anexo 2; disponível na versão on-line do JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48).(29)
O objetivo do presente estudo foi avaliar como os TS são realizados e interpretados nos centros que aceitaram par-ticipar do estudo e que realizam esses testes rotineiramente em diversas cidades do estado de SP, comparando sua rotina àquelas especificadas em diretrizes internacionais. Ao todo, esses centros realizam aproximadamente 4.500 testes por ano.
MÉTODOSTrata-se de um estudo observacional descritivo de coorte transversal. Um total de 18 centros que realizam rotineiramente os TS no estado de SP foi convidado a participar do estudo em 2013. O estudo foi aprovado pelo Comitê de Ética em Pesquisa da Universidade Estadual de Campinas (Protocolo nº 86624/2012) e foi realizado de acordo com a Declaração de Helsinki. Todos os participantes assinaram um termo de consentimento livre e esclarecido.
Foi desenvolvido um questionário constituído por 54 questões que incluíam todas as etapas da realização dos TS: estimulação do suor, coleta do suor e dosagem de Cl−(12,13) (Anexo 3; disponível na versão on-line do JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48). O questionário foi entregue ao pessoal responsável pela realização dos TS em todos os centros incluídos no estudo. Em nosso estudo, dois pesquisadores realizaram as entrevistas simultaneamente. Os dados foram compilados em planilhas Excel, e os resultados foram apresentados em tabelas e figuras.
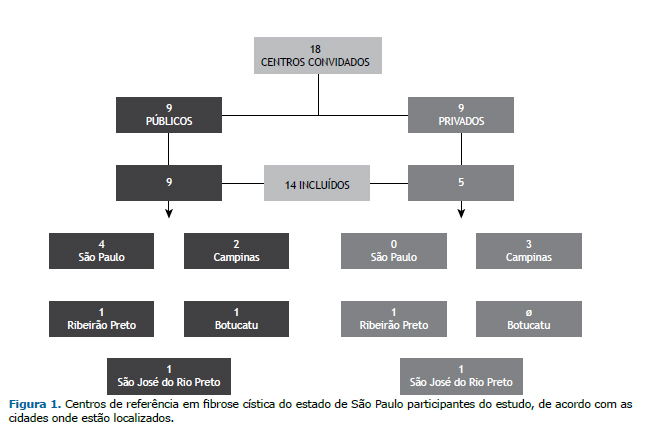
RESULTADOSIdentificou-se um total de 18 centros que realizam TS como parte de sua rotina (9 privados e 9 públicos). Dos 18 centros, 14 aceitaram participar do estudo, sendo 5 (35,7%) instituições privadas e 9 (64,3%) centros públicos de saúde (Figura 1). No momento da visita, 4 dos centros não estavam realizando TS por falta de insumos. Os 14 centros incluídos no estudo realizam aproximadamente 4.500 TS/ano, sendo que 4.000 desses testes são realizados em centros públicos. O número de TS/ano por centro é apresentado na Figura 2. Entre os 14 centros, o tempo de experiência na realização de TS variou de 1 a > 20 anos (Anexo 4; disponível na versão on-line do JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48).

Entrevistamos os profissionais envolvidos na realização dos três estágios dos TS nos centros participantes. Quanto à ocupação, um era médico, sete eram biomédicos, dois eram biólogos, quatro eram enfermeiros, e sete eram técni-cos de enfermagem. Entre os 14 centros, 11 profissionais foram treinados por colegas do mesmo centro (treinamento interno), 2 foram treinados em outro centro (treinamento externo), e 1 recebeu treinamento tanto interno quanto externo. Apenas 1 dos centros conhecia as diretrizes para TS e possuía uma versão impressa do manual de proce-dimentos operacionais padrão.
Entre os 14 centros, 2 não possuíam equipamentos para estimulação do suor. Alguns centros utilizavam mais de um dispositivo, sendo os equipamentos utilizados nove MSCS; um CF-Indicator® (Polychrome Medical Inc., Brooklyn Center, MN, EUA); cinco Iontokit® (Advanced Instruments Inc., Norwood, MA, EUA); e um produzido pela Qualiterm (Qualiterm, São Paulo, Brasil). Os cinco dispositivos conectados à rede elétrica eram os seguintes: três Iontokit® (Advanced Instruments Inc.), um Iontoplus® (NAIMCO Inc., Chattanooga, TN, EUA); e um aparato artesanal. Esses dispositivos eram mais velhos, com tempo médio de uso de 15 anos (Figura 3).
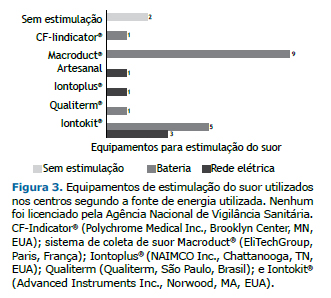
Ao todo, havia 21 equipamentos de estimulação em 12 dos centros. Nenhum dos equipamentos tinha registro ofici-al para o diagnóstico clínico de FC no país. Entre esses, havia 8 MSCS, dos quais 7 não estavam em operação por falta de peças sobressalentes ou insumos ou porque quebraram menos de um ano atrás. Entre os 12 centros que realizavam a estimulação do suor, 8 utilizavam o antebraço/braço para a colocação dos eletrodos, enquanto os outros 4 utilizavam outros locais (Figura 4A). A estimulação do suor era conseguida por meio de uma ou mais das seguintes técnicas: uso de cobertor, em 6 centros; uso de casaco, em 6; corrida ou caminhada ao ar livre, em 4; oclusão da pele com filme plástico, em 2; uso de curativo, em 1; e uso de parafilme, em 1. O suor era coletado do paciente com o auxílio de uma colher descartável e colocado em um tubo estéril para a dosagem de Cl− em 1 centro. Em 2 centros privados, o suor era induzido sem estimulação por dispositivos de iontoforese (Anexo 5; disponível na versão on-line do JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48).
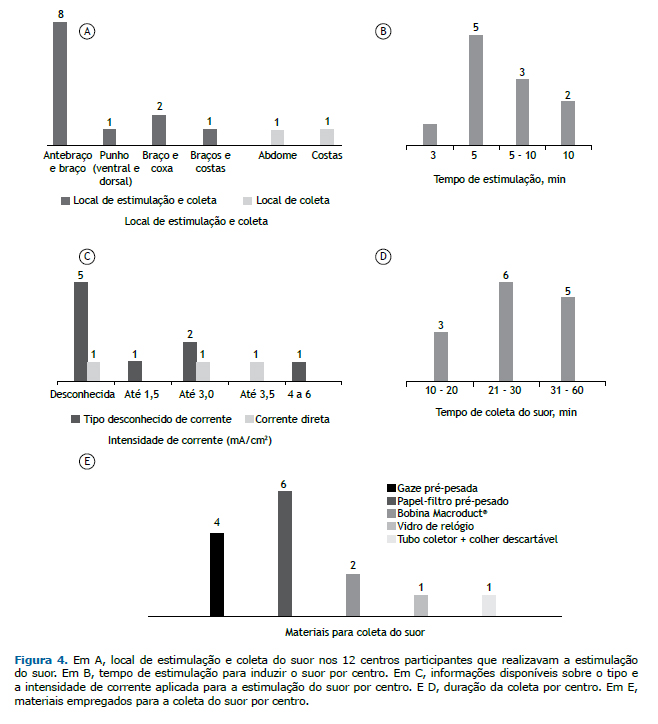
O tempo de estimulação elétrica para a indução do suor variou de 3 a 10 min (Figura 4B). O tipo de corrente utili-zado era conhecido em apenas 3 dos 14 centros (corrente direta) e desconhecido em 9, dos quais 5 também não informaram a intensidade de corrente utilizada. A intensidade de corrente era conhecida em 5 centros (Figura 4C). Em 4 centros (28,6%), foi relatado que o procedimento de estimulação elétrica havia causado queimadura de pele em alguns pacientes; porém, 5 centros (35,7%) relataram que isso nunca havia acontecido com nenhum de seus pacientes, enquanto 3 centros (21,4%) não puderam relatar essa informação, pois os entrevistados estavam traba-lhando há pouco tempo nesses centros. Finalmente, 2 centros (14,3%) não utilizavam estimulação elétrica.
O tempo de coleta do suor variou de 10 a 60 min (Figura 4D). Os materiais empregados para a coleta do suor nos 14 centros são descritos na Figura 4E. Cabe ressaltar que 2 centros estavam utilizando materiais/técnicas incomuns: um vidro de relógio era colocado sobre o local da estimulação com pilocarpina e então fixado com fita, e as gotículas de suor eram coletadas utilizando uma pipeta estéril ou uma colher descartável e colocadas em um tubo estéril em 1 dos centros, enquanto o outro centro não tinha balanças de precisão apropriadas para a pesagem do material coleta-do. Além disso, 2 centros realizavam a estimulação do suor sem utilizar nenhum tipo de equipamento e, portanto, foram desconsiderados quanto a essa questão.
A dosagem de Cl− era realizada por meio da técnica de titulação manual e do teste quantitativo coulométrico (clo-ridrômetro), em 6 centros cada. Entre estes últimos, 1 centro não estava em operação. A necessidade de repetição do TS deveu-se a suor insuficiente ou a causas desconhecidas em 10 e em 4 centros, respectivamente. A taxa de repeti-ção dos TS foi de 5%, em 1 centro; de 10-20%, em 7; de 30%, em 1; e > 50%, em 1.
O número de pessoas que coletavam o suor e realizavam os TS está resumido na Figura 5A. O mesmo profissional era responsável pela coleta e realização do TS em 6 centros. A quantidade mínima aceitável de suor para a realiza-ção de um teste do suor confiável variou consideravelmente entre os centros e está resumida na Figura 5B. Em 11 centros que utilizavam papel-filtro ou gaze para a coleta do suor, a quantidade de suor variou consideravelmente de 50 a 100 mg, e 1 centro não tinha conhecimento do valor aceitável (Figura 5B). Quanto aos 3 centros que utilizavam o MSCS, o volume aceitável de suor era de 15 µl, 20 µl e sem padrão. O centro que utilizava o vidro de relógio rela-tou que o volume mínimo necessário era de 20 µl. A maioria dos profissionais dos centros desconhecia o número correto de testes com resultados positivos (Cl− ≥ 60 mEq/l) para o diagnóstico definitivo de FC (Figura 5C). A Figura 6 mostra o quão aderente às diretrizes da Cystic Fibrosis Foundation(13) era cada um dos 14 centros.

 DISCUSSÃO
DISCUSSÃONo Brasil, a diversidade de expressão da doença é condicionada pela miscigenação, que aumenta muito a diversi-dade genética expressa em uma variabilidade de mutações no gene CFTR em nossa população, tornando assim o diagnóstico genético difícil e dispendioso. O acompanhamento de pacientes com FC no Brasil é realizado em centros de referência, a maioria dos quais é público e vinculado ao Sistema Único de Saúde com apoio financeiro público e geralmente associada a universidades. O estado de SP possui a segunda renda per capita do país e o maior número de centros de referência em FC (n = 7), todos os quais são centros públicos de saúde (Anexo 6; disponível na versão on-line do JBP; http://www.jornaldepneumologia.com.br/detalhe_anexo.asp?id=48).
O presente estudo sobre a realização do TS em 14 centros, que, ao todo, realizam 4.500 testes/ano, revelou que não há uniformidade nos procedimentos do TS e que há sérias dificuldades em sua realização e condições significativamente inadequadas, que o deixam muito distante das diretrizes internacionalmente aceitas.
Estudos semelhantes, no entanto, já haviam sido realizados sobre a qualidade dos TS em diversos centros em vá-rios outros países, também apresentando diversidade significativa e resultados inconsistentes. (12,30,31) A confirmação diagnóstica da FC possibilita que os centros de saúde proporcionem melhores cuidados e acompanhamento, o que se traduz em maior expectativa de vida para os pacientes.(8) Isso também foi encontrado em nosso centro de referência.(32) A padronização dos TS é fundamental para um diagnóstico confiável de FC.
Em particular, o presente estudo mostrou que não há uniformidade na realização do TS em seus três estágios (estimulação, coleta e quantificação) nos centros participantes. As principais questões e possíveis alternativas para as três etapas podem ser resumidas da seguinte forma:
(i) Estimulação: o desconhecimento da existência de equipamentos adequados para estimulação do suor e do uso dos mesmos estava presente em aproximadamente 30% dos centros participantes. Métodos alternativos de es-timulação, não aderentes às diretrizes internacionais (e.g., exposição ao sol com um cobertor, exposição ao sol dentro de um carro, uso de equipamentos de indução do suor não certificados e falta de conhecimento de técni-cas de estimulação), eram praticados em 4 dos centros e podem até prejudicar a saúde dos pacientes (e.g., queimadura de pele, desidratação ou até morte). Além disso, esses 4 centros também coletavam amostras de suor inadequadas para dosagem de Cl−. Cada centro deve contar com a técnica de estimulação por iontoforese com pilocarpina, utilizar equipamentos certificados de estimulação e submeter os mesmos a manutenção e ca-libração regulares pelo fabricante ou por uma empresa qualificada a fim de garantir a segurança do procedi-mento. Cada centro também deve proporcionar (ou buscar em outro lugar) treinamento adequado dos profissi-onais responsáveis pelo manuseio dos equipamentos.
(ii) Coleta: a maioria dos centros participantes utilizava métodos para coleta do suor que estavam de acordo com as recomendações estabelecidas (uso de papel-filtro, gaze ou MSCS). No entanto, 2 centros utilizavam métodos de coleta alternativos e não recomendados (uso de uma colher ou um vidro de relógio), que afetam a confiabi-lidade do TS. Embora o MSCS seja descrito como um método adequado para a coleta do suor pelo Departa-mento de Saúde dos EUA desde 2003, o mesmo não estava registrado no Brasil durante o período estudado.(13) Em setembro de 2014, o uso do MSCS foi devidamente licenciado, bem como o do cloridrômetro digital, o que possibilitou a manutenção periódica adequada dos equipamentos e a aquisição ininterrupta de insumos. Esse fato possivelmente mudará o cenário atual: 7 centros não estavam utilizando o MSCS por falta de peças so-bressalentes/insumos, dificuldades na manutenção dos equipamentos ou falta de profissionais treinados capa-zes de utilizar o sistema (como realizar a indução do suor, a coleta do suor e a dosagem de Cl−). Outro fator a ser considerado é o alto custo dos insumos, que são importados, em comparação ao método tradicional de es-timulação e coleta para o método de GC, que utiliza pilocarpina e papel-filtro ou gaze para a coleta do suor.
(iii) Quantificação: A dosagem de Cl− deve ser quantitativa e realizada por coulometria, fotometria de chama ou titulação manual. Independentemente do procedimento, não houve problemas quanto a essa questão nos cen-tros participantes, exceto pelo fato de que a condutividade, que não é um procedimento confiável, foi utilizada para o diagnóstico da FC em 2 centros. Em outro centro, a condutividade foi utilizada apenas para triagem, o que é uma abordagem apropriada e correta.
A FC é uma doença progressiva, que exige que os pacientes sejam atendidos em centros de referência para que recebam os melhores cuidados de saúde e tratamento adequado. Um diagnóstico seguro e confiável é o primeiro passo, e é fundamental para a orientação dos pacientes e suas famílias pela equipe médica.
O presente estudo mostra a real situação dos TS no estado de SP, a qual pode ser representativa da situação geral para TS no Brasil. Ao todo, nossos resultados mostram que, nos centros participantes, os TS estão muito distantes das diretrizes internacionalmente aceitas. Há necessidade urgente de equipamentos domésticos ou importados para estimulação do suor e para dosagem de Cl− no suor de acordo com as diretrizes internacionais. A manutenção deve ser adequada, e peças sobressalentes e insumos devem estar sempre disponíveis para que os resultados obtidos sejam confiáveis e apropriados.
A partir desse momento único, quando comemoramos a introdução da TN para FC em todos os estados do Brasil, devemos estar preparados para superar os desafios à nossa frente. Esses desafios só podem ser superados se forem primeiramente identificados e enfrentados de forma objetiva pelas equipes de FC. Além disso, precisamos trabalhar juntos para mudar a realidade atual; acreditamos que só poderemos construir uma nova realidade se primeiramente nos conscientizarmos das limitações e dificuldades inerentes a cada centro individual.
O presente estudo serve como um alerta sobre a necessidade de padronização dos TS no Brasil por meio da cons-trução e adoção de diretrizes para o diagnóstico da FC. A questão tem estado na agenda de discussões do Grupo Brasileiro de Estudos em Fibrose Cística.
Os diretores dos centros de referência em FC devem revisar cada etapa atual da realização dos TS, trabalhando bem de perto com o pessoal de laboratório, o que sem dúvida melhorará a qualidade dos resultados de forma a proporcionar um diagnóstico confiável de FC e minimizar o possível viés dos TS. Os profissionais que realizam os TS devem conhecer as especificidades da doença e reconhecer o importante papel de um TS corretamente realizado para a confirmação ou exclusão do diagnóstico de FC. Tal treinamento deve ser proporcionado pelos centros de referência a seus profissionais, fomentando a participação dos mesmos em treinamentos internos ou externos, even-tos científicos e outros fóruns de discussão. A proximidade entre os diferentes profissionais que trabalham na área da FC, bem como a troca de informações entre os diferentes centros de referência, permitirá aumentar o conhecimento sobre os TS e, portanto, melhorar os procedimentos neles envolvidos para um diagnóstico mais confiável de FC.
Em conclusão, nos vários centros participantes do estado de SP, não encontramos uniformidade nas etapas execu-tadas na realização dos TS e encontramos grande afastamento das diretrizes internacionalmente aceitas para TS. As principais condições inadequadas incluíram a produção insuficiente de suor, a falta de equipamentos de estimulação ou de equipamentos de química clínica, a ausência de protocolos escritos e o uso de equipamentos de medição não certificados e obsoletos. Embora saibamos que há diversas dificuldades e barreiras a serem superadas, precisamos nos movimentar no sentido de atender os 23 tópicos das diretrizes da Cystic Fibrosis Foundation(13) para que os TS sejam realizados adequadamente para esse fim. Há necessidade urgente de padronização dos TS, de treinamento de pessoal qualificado, de equipamentos adequados e de certificação. Essas são condições essenciais para um diagnós-tico confiável de FC, especialmente com a crescente demanda resultante da TN em todo o país, e para a avaliação do possível benefício clínico com o uso de moduladores de CFTR.
AGRADECIMENTOSGrupo Colaborativo de Estudos em Fibrose Cística: Francisco Ubaldo Vieira Junior, Margarida Duarte Amaral, Neiva Damaceno, Joaquim Carlos Rodrigues, Giesela Fleischer Ferrari, Vera Lucia Sdepanian, João Batista Salomão Junior, Sônia Mayumi Chiba, Sônia Letícia Silva Lorena e Maria Inêz Machado Fernandes.
REFERÊNCIAS1. Levy H, Farrell PM. New challenges in the diagnosis and management of cystic fibrosis. J Pediatr. 2015;166(6):1337-41. https://doi.org/10.1016/j.jpeds.2015.03.042
2. Cystic Fibrosis Data Network [homepage on the Internet]. Belconnen, Australia: Geoff Sims Consulting Pty Ltd.; c2013 [cited 2016 Feb 2]. Cystic fibrosis data. Available: http://www.cysticfibrosisdata.org/Home.htm
3. Raskin S, Pereira-Ferrari L, Reis FC, Abreu F, Marostica P, Rozov T, et al. Incidence of cystic fibrosis in five different states of Brazil as determined by screening of p.F508del, mutation at the CFTR gene in newborns and patients. J Cyst Fibros. 2008;7(1):15-22. https://doi.org/10.1016/j.jcf.2007.03.006
4. Camargos P, Gomes DL, Alvim CG, Gomes FS, Cajazeiro JM. From lip to lab: salty tasting skin is the main clue that raises clinical suspicion of cystic fibrosis in young infants. Acta Paediatr. 2015;104(5):e210-5. https://doi.org/10.1111/apa.12958
5. Dijk FN, Fitzgerald DA. The impact of newborn screening and earlier intervention on the clinical course of cystic fibrosis. Paediatr Respir Rev. 2012;13(4):220-5. https://doi.org/10.1016/j.prrv.2012.05.003
6. Vernooij-van Langen AM, Gerzon FL, Loeber JG, Dompeling E, Dankert-Roelse JE. Differences in clinical condition and genotype at time of diagnosis of cystic fibrosis by newborn screening or by symptoms. Mol Genet Metab. 2014;113(1-2):100-4. https://doi.org/10.1016/j.ymgme.2014.07.012
7. van der Ploeg CP, van den Akker-van Marle ME, Vernooij-van Langen AM, Elvers LH, Gille JJ, Verkerk PH, et al. Cost-effectiveness of newborn screening for cystic fibrosis determined with real-life data. J Cyst Fibros. 2015;14(2):194-202. https://doi.org/10.1016/j.jcf.2014.08.007
8. Smyth AR, Bell SC, Bojcin S, Bryon M, Duff A, Flume P, et al. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J Cyst Fibros. 2014;13 Suppl 1:S23-42. https://doi.org/10.1016/j.jcf.2014.03.010
9. Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics. 1959;23(3):545-9.
10. Collie JT, Massie RJ, Jones OA, LeGrys VA, Greaves RF. Sixty-five years since the New York heat wave: advances in sweat testing for cystic fibrosis. Pediatr Pulmonol. 2014;49(2):106-17. https://doi.org/10.1002/ppul.22945
11. Cystic Fibrosis Foundation [homepage on the Internet]. Bethesda: the Foundation; [cited 2016 Feb 2]. Available: https://www.cff.org/
12. Cirilli N, Padoan R, Raia V; ICFS Sweat Test Working Group. Audit of sweat testing: a first report from Italian Cystic Fibrosis Centres. J Cyst Fibros. 2008;7(5):415-22. https://doi.org/10.1016/j.jcf.2008.03.005
13. LeGrys VA, Yankaskas JR, Quittell LM, Marshall BC, Mogayzel PJ Jr; Cystic Fibrosis Foundation. Diagnostic sweat testing: the Cystic Fibrosis Foundation guidelines. J Pediatr. 2007;151(1):85-9. https://doi.org/10.1016/j.jpeds.2007.03.002
14. Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153(2):S4-S14. https://doi.org/10.1016/j.jpeds.2008.05.005
15. Castellani C, Southern KW, Brownlee K, Dankert Roelse J, Duff A, Farrell M, et al. European best practice guidelines for cystic fibrosis neonatal screening. J Cyst Fibros. 2009;8(3):153-73 https://doi.org/10.1016/j.jcf.2009.01.004
16. Lezana JL, Vargas MH, Karam-Bechara J, Aldana RS, Furuya ME. Sweat conductivity and chloride titration for cystic fibrosis diagnosis in 3834 subjects. J Cyst Fibros. 2003;2(1):1-7. https://doi.org/10.1016/S1569-1993(02)00146-7
17. US National Committee for Clinical Laboratory Standards. Sweat testing: sample collection and quantitative analysis; approved guideline. 2nd ed. Waynes (PA): US National Committee for Clinical Laboratory Standards; 2000 Jun. Document No: C34-A2.
18. US National Committee for Clinical Laboratory Standards. Sweat testing: sample collection and quantitative analysis; approved guideline. 3rd ed. Waynes (PA): US National Committee for Clinical Laboratory Standards; 2009 Dec. Document No: C34-A3.
19. LeGrys VA. Sweat analysis proficiency testing for cystic fibrosis. Pediatr Pulmonol. 2000;30(6):476-80. https://doi.org/10.1002/1099-0496(200012)30:6<476::AID-PPUL7>3.0.CO;2-O
20. LeGrys VA. Assessment of sweat-testing practices for the diagnosis of cystic fibrosis. Arch Pathol Lab Med. 2001;125(11):1420-4.
21. Baumer JH. Evidence based guidelines for the performance of the sweat test for the investigation of cystic fibrosis in the UK. Arch Dis Child. 2003;88(12):1126-7. https://doi.org/10.1136/adc.88.12.1126
22. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663-72. https://doi.org/10.1056/NEJMoa1105185
23. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220-31. https://doi.org/10.1056/NEJMoa1409547
24. Quinton P, Molyneux L, Ip W, Dupuis A, Avolio J, Tullis E, et al. -adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am J Respir Crit Care Med. 2012;186(8):732-9. https://doi.org/10.1164/rccm.201205-0922OC
25. Gonska T, Ip W, Turner D, Han WS, Rose J, Durie P, et al. Sweat gland bioelectrics differ in cystic fibrosis: a new concept for potential diagnosis and assessment of CFTR function in cystic fibrosis. Thorax. 2009;64(11):932-8. https://doi.org/10.1136/thx.2009.115295
26. Sousa, M, Servidoni MF, Vinagre A, Ramalho AS, Bonadia LC, Felício V, et al. Measurements of CFTR-mediated Cl- secretion in human rectal biopsies constitute a robust biomarker for Cystic Fibrosis diagnosis and prognosis. PLoS One. 2012;7(10):e47708. https://doi.org/10.1371/journal.pone.0047708
27. Gonçalves AC, Marson FA, Mendonça RM, Ribeiro JD, Ribeiro AF, Paschoal IA, et al. Saliva as a potential tool for cystic fibrosis diagnosis. Diagn Pathol. 2013;8:46. https://doi.org/10.1186/1746-1596-8-46
28. Ng RT, Marson FA, Ribeiro JD, Ribeiro AF, Bertuzzo CS, Ribeiro MA, et al. Nasal potential difference in cystic fibrosis considering severe CFTR mutations. Dis Markers. 2015;2015:306825
29. Instituto Brasileiro de Geografia e Estatística [homepage on the Internet]. São Paulo: IBGE; c2016 [cited 2016 Feb 2]. Estados@--São Paulo: Botucatu. [about 3 screens]. Available from: http://www.ibge.gov.br/estadosat/perfil.php?sigla=sp
30. Kirk JM. Inconsistencies in sweat testing in UK laboratories. Arch Dis Child. 2000;82(5):425-7. https://doi.org/10.1136/adc.82.5.425
31. Mackay R, George P, Kirk J. Sweat testing for cystic fibrosis: A review of New Zealand laboratories. J Paediatr Child Health. 2006;42(4):160-4. https://doi.org/10.1111/j.1440-1754.2006.00822.x
32. MARSON FA, HORTENCIO TD, AGUIAR KC, RIBEIRO JD; CYFIUC GROUP. DEMOGRAPHIC, CLINICAL, AND LABORATORY PARAMETERS OF CYSTIC FIBROSIS DURING THE LAST TWO DECADES: A COMPARATIVE ANALYSIS. BMC PULM MED. 2015;15:3. HTTPS://DOI.ORG/10.1186/1471-2466-15-3











 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket