ABSTRACT
Objective: To evaluate the association between nutritional status measurements and pulmonary function in children and adolescents with cystic fibrosis. Methods: We evaluated the nutritional status of 48 children and adolescents (aged 6-18 years) with cystic fibrosis based on body mass index (BMI) and body composition measurements-mid-arm muscle circumference (MAMC) and triceps skinfold thickness (TST)-at a referral center in the city of Rio de Janeiro, Brazil. Pulmonary function was assessed by means of spirometry, using FEV1 to classify the severity of airway obstruction. We used Student's t-tests for comparisons between proportions and linear regression analysis for associations between continuous variables. The level of significance was set at p < 0.05. Results: The evaluation of nutritional status based on BMI identified a smaller number of malnourished patients than did that based on MAMC (14 vs. 25 patients, respectively). Most of the patients presented mild pulmonary disease. Mean FEV1 was 82.5% of predicted. Pulmonary function was found to correlate significantly with BMI, MAMC and TST (p = 0.001, p = 0.001 and p = 0.03, respectively). All subjects with moderate or severe pulmonary involvement were considered malnourished based on BMI and body composition parameters. Of the 25 patients considered malnourished based on body composition (MAMC), 19 were considered well-nourished based on their BMI. Conclusions: In the present study, all nutritional status measurements correlated directly with the pulmonary function of children and adolescents with cystic fibrosis. However, body composition measurements allowed earlier detection of nutritional deficiencies.
Keywords:
Cystic fibrosis; Body composition; Spirometry; Nutrition assessment.
RESUMO
Objetivo: Avaliar a associação de medidas do estado nutricional com a função pulmonar de crianças e adolescentes com fibrose cística. Métodos: Foi avaliado o estado nutricional pelo índice de massa corpórea (IMC) e por medidas de composição corporal-circunferência muscular do braço (CMB) e medida da dobra cutânea triciptal (DCT)-de 48 crianças e adolescentes (6-18 anos) com fibrose cística em um centro de referência na cidade do Rio de Janeiro. A função pulmonar foi analisada por espirometria, e o parâmetro adotado para a classificação da obstrução das vias aéreas foi VEF1. Utilizou-se o teste t de Student para a comparação entre proporções e a regressão linear para associação entre variáveis contínuas. O nível de significância adotado foi p < 0,05. Resultados: A avaliação do estado nutricional através do IMC encontrou menos pacientes desnutridos do que a realizada através da CMB (14 vs. 25, respectivamente) A maioria dos pacientes apresentou doença pulmonar leve. A média do VEF1 foi de 82,5% do previsto. Houve associação do IMC, da CMB e da DCT com a função pulmonar (p = 0,001, p = 0,001 e p = 0,03, respectivamente). Todos os pacientes com comprometimento moderado e grave pulmonar eram desnutridos pela composição corporal (CMB). Entre os 25 pacientes considerados desnutridos através da composição corporal (CMB), 19 apresentavam peso adequado quando avaliados pelo IMC. Conclusões: Todas as medidas do estado nutricional apresentaram associação direta com a função pulmonar das crianças e adolescentes com fibrose cística no estudo. Entretanto, as medidas de composição corporal revelaram, de forma mais precoce, possíveis deficiências nutricionais.
Palavras-chave:
Fibrose cística; Composição corporal; Espirometria; Avaliação nutricional.
IntroductionCystic fibrosis (CF) is an autosomal recessive genetic disease characterized by a wide variety of clinical manifestations, chief among which are progressive suppurative COPD, pancreatic insufficiency, secondary malnutrition, increased sodium and chloride concentrations in sweat and male infertility in adulthood.(1)
Chronic bacterial infection of the respiratory tract causes destruction and loss of pulmonary function, and respiratory disease is one of the leading causes of decreased longevity and of the increased frequency of hospital admissions among such patients.(2)
Maintaining adequate nutritional status is essential for the integrity of the respiratory system in CF.(3,4) One of the leading causes of nutritional depletion is increased energy expenditure due to pulmonary inflammation and infection,(3) characterizing the direct relationship between nutritional status and pulmonary function.
Mineral, electrolyte and energy disturbances at the muscle level are
responsible for the dysfunction of and decrease in accessory respiratory muscle and diaphragm contractility. Malnutrition can affect the parenchyma, the pulmonary immune response and the respiratory drive.(5) This profile results in decreased respiratory performance on exertion and increased susceptibility to pulmonary infections, as well as in respiratory failure.(6)
A study on protein turnover in adults suggested that individuals with CF are in a chronic state of catabolic stress or malnutrition, or both, related to recurrent pulmonary exacerbations that adversely affect energy balance and protein metabolism.(7)
In recent years, the importance of nutrition in the evolution of CF has been cited, in many studies, as being essential for determining the prognosis of the disease.(4,5,7,8)
Including the use of anthropometric criteria, in order to quantify and qualify nutritional status more precisely, is essential for timely diagnosis and early appropriate intervention in changes or alterations in nutritional status.
The objective of the present study was to evaluate the association between nutritional status, determined based on anthropometric and body composition indices, and pulmonary function in children and adolescents with CF treated at a referral center in the city of Rio de Janeiro.
MethodsThis was a cross-sectional descriptive study involving 48 children and adolescents with CF, according to the diagnostic criteria established in the Cystic Fibrosis Foundation consensus.(9) The study inclusion criteria were as follows: being between 6 and 18 years of age; and being under treatment at the Nutrition Outpatient Clinic of the Oswaldo Cruz Foundation Instituto Fernandes Figueira (IFF, Fernandes Figueira Institute). Patients with diabetes or liver disease were excluded. The research project was approved by the IFF Research Ethics Committee. The legal guardians of all participating patients gave written informed consent.
Anthropometric and respiratory function data were colleted between January of 2006 and January of 2008.
Weight (in kg) was measured using an anthropometric platform scale (Filizola S.A., São Paulo, Brazil), and height (in cm) was measured using a stadiometer attached to the scale. Patients were wearing no shoes and little clothing, and their heads were positioned with the Frankfurt plane parallel to the floor. Body mass index (BMI) was calculated by dividing weight in kilograms by height in meters squared (kg/m2). Arm circumference (AC) was measured at the midpoint between the scapula acromion and the ulna olecranon of the right arm, which was relaxed. Triceps skinfold thickness (TST; in mm) was measured at the midpoint of the posterior face of the right arm using a Lange® skinfold caliper (Cambridge Scientific Industries, Cambridge, MD, USA). The measurements were performed in triplicate, and the mean value of the three measurements was used for analysis.
Mid-arm muscle circumference (MAMC) was calculated by subtracting the value obtained by multiplying TST (converted to cm) by ω from AC (cm), according to the following equation(10):
MAMC(cm) = AC(cm) − (TST(cm) × ω)
The population reference for nutritional assessment was the graph developed by the National Center for Health Statistics in 2000.(11)
Nutritional assessment data were analyzed in percentiles, in accordance with the Consensus Report on Nutrition for Pediatric Patients with Cystic Fibrosis.(7) Patients presenting BMI for age (BMI/A) below the 10th percentile were considered malnourished, those presenting BMI/A between the 10th and 25th percentiles were considered to be at nutritional risk, and those presenting BMI/A above the 25th percentile were considered well-nourished. Low height was defined as height-for-age below the 5th percentile.(7)
The MAMC and TST values were determined using the parameters established by Frisancho,(10) values below or at the 5th percentile being considered low. Pubertal stage was assigned using Tanner staging.(12)
Pulmonary function was assessed at the IFF Pulmonary Function Laboratory. To ensure the validity of the results, the minimum age for participants was set at 6 years, since spirometry requires patient cooperation. We used FEV1 to classify pulmonary impairment in terms of obstruction. The technique used to perform the tests, as well as the reference values, were in accordance with the recommendations of the American Thoracic Society, which classifies predicted percentage values as follows: mild obstruction, ≥ 70%; moderate obstruction, 60-69%; moderately severe obstruction, 50-59%; severe obstruction, 35-49%; extremely severe obstruction, < 35%.(13) Overweight patients were excluded.
Data were analyzed using the program Statistical Package for the Social Sciences, version 8.0 for Windows (SPSS Inc., Chicago, IL, USA). We used Student's t-tests for comparisons between proportions and linear regression analysis for associations between continuous variables. The level of significance was set at p < 0.05.
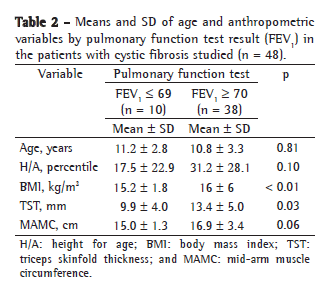
ResultsOf the 48 patients studied, 29 were female and 19 were male. The mean age was 10.8 ± 3.3 years (Table 1). Most of the individuals (79%) presented mild pulmonary function impairment (Table 2).
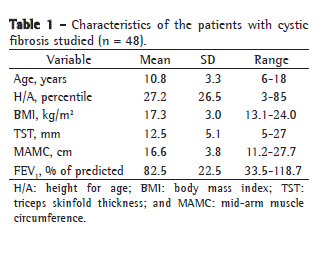
None of the patients presented decreased fat deposition based on TST. The prevalence of low height in the group was 19.6%, and none of the patients presented delayed puberty.
Nutritional assessment based on MAMC revealed a higher percentage of malnutrition (52%) than did that based on BMI/A, which revealed that 31.2% of the participants were malnourished and 14.5% were at nutritional risk (Table 3). Of the patients considered malnourished based on MAMC, 24% were classified as being at nutritional risk and 39% were classified as well-nourished based on BMI/A.
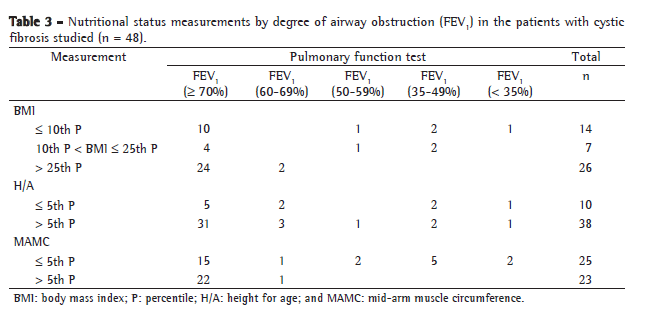
All patients with moderate or severe airway obstruction were considered malnourished based on MAMC (Table 3).
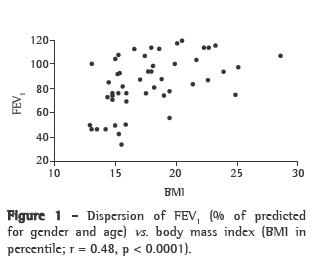
Although the degree of pulmonary impairment presented a statistically significant association with BMI and with TST (p = 0.001 and p = 0.03, respectively), it presented only a tendency toward an association with MAMC (p = 0.06; Table 2). Linear regression revealed a moderate relationship between BMI and FEV1 (r = 0.46; p = 0.00001) and between FEV1 and MAMC (r = 0.46; p = 0.0001; Figures 1 and 2).
 Discussion
DiscussionThe present study found that the prevalence of malnutrition based on MAMC was higher that that based on BMI/A, although the latter presented a higher malnutrition cut-off point for patients with CF (10th percentile) than for the general population (5th percentile). Of the patients considered malnourished based on MAMC, 39% were considered normal based on BMI/A. These data reveal the relevance of analyzing body composition to assess nutritional status more accurately.
The moderate relationship found between nutritional status and FEV1 (Figures 1 and 2) in the patients in our sample is probably due to the fact that decreased pulmonary function cannot be explained solely by increased impairment of nutritional status. Noncompliance with treatment and noncompliance with regular respiratory therapy are examples of other factors that destabilize pulmonary function.
The mild impairment of pulmonary function presented by most of the patients in our sample might have played a role in the tendency (p < 0.06) toward an association between pulmonary function and nutritional status assessment based on MAMC. In cases of mild pulmonary impairment, nutritional depletion occurs due to other factors, such as the type of genetic mutation, pancreatic insufficiency, loss of bile salts and acids, gastroesophageal reflux, intestinal resection and the emotional status of patients. Therefore, the pathophysiology of malnutrition found in the present study is similar to that of primary malnutrition, which retains muscle mass at the expense of fat mass.
Adiposity presented a statistically significant association with pulmonary function (p = 0.03), although the TST measurements of all patients were within the range of normality (Table 2). It is possible that the TST cut-off point (5th percentile) should be higher for patients with CF. One group of authors also found a positive association between adiposity and pulmonary function after having studied the body composition of 50 pediatric patients with CF by means of dual-energy X-ray absorptiometry.(6)
All patients with moderate or severe airway obstruction were considered malnourished based on MAMC (Table 3). This result was expected, since there is a consensus in the literature that pulmonary inflammation and infection increase energy expenditure and are some of the leading causes of nutritional depletion, especially of muscle mass, even in patients whose food intake is adequate or above that recommended.(2,5,14,15)
The low height found in the present study could be attributed to genetics. However, the lack of a data record for the determination of the genetic target precludes such an assumption. Studies involving Brazilian and American children found similar results and showed that low height in patients with CF does not depend on the level of development of the country or on the genetic load but is related to factors inherent to the pathophysiology of the disease.(16,17)
The age-related decline in pulmonary function is universal in CF.(18) However, in the present study, the mean age of the patients was similar in the two groups divided based on the degree of pulmonary impairment.
In our study, 10 patients (19.6%) presented low height, and, of those, 60% presented moderate or severe pulmonary function impairment. This result is in accordance with data from a report by the Cystic Fibrosis Foundation,(19) in which height-for-age indices were found to be below the 5th percentile in 20% of children with CF. It has long been recognized that nutritional deficiency can significantly affect pulmonary function, as well as that chronic pulmonary disease can lead to growth deficit and delayed development, creating a vicious cycle between malnutrition and respiratory disorders.(14,20)
Various studies have demonstrated that BMI is a more sensitive parameter than is weight for height.(4,17,21,22) In the present study, BMI presented a statistically significant association with pulmonary function.
However, BMI should not be used in isolation to assess the nutritional status of patients with CF, since it does not determine body composition.
In CF, the imbalance between protein synthesis and protein degradation to meet the energy requirements of the body can lead to a disproportionate loss of lean body mass, with muscle atrophy, including respiratory muscle atrophy.(18) Therefore, a change in body composition can lead to malnutrition and decreased pulmonary function.(6,7,23)
Since body weight is composed primarily of fat mass and fat-free mass, observing these two body compartments separately can provide more accurate information about nutritional status, which is directly related to pulmonary disease-the leading cause of morbidity and mortality in CF.
The results of the present study show that the change in body composition occurs prior to the decrease in weight and height in children and adolescents with CF. Since the assessment of nutritional status, including the measurement of fat and muscle reserves based on TST and AC, is an inexpensive and easily performed method, health professionals should be encouraged to use it during routine visits, identifying patients at risk of malnutrition earlier, so that the nutritional intervention can be immediately recommended, and possibly delaying the progression of the pulmonary disease.
In the present study, nutritional status measurements correlated with pulmonary function in children and adolescents with CF. However, body composition measurements allowed earlier detection of nutritional deficiencies. Therefore, the combination of anthropometric measurements (BMI) and body composition measurements increases accuracy in the nutritional diagnosis and detects early changes in nutritional status, informing decisions regarding nutritional interventions and delaying the decline in pulmonary function in such patients.
References 1. Rozov T, Cunha MT, Nascimento O, Quittner AL, Jardim JR. Linguistic validation of cystic fibrosis quality of life questionnaires. J Pediatr (Rio J). 2006;82(2):151-6.
2. Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med. 1996;154(5):1229-56.
3. Peterson ML, Jacobs DR Jr, Milla CE. Longitudinal changes in growth parameters are correlated with changes in pulmonary function in children with cystic fibrosis. Pediatrics. 2003;112(3 Pt 1):588-92.
4. Stapleton D, Kerr D, Gurrin L, Sherriff J, Sly P. Height and weight fail to detect early signs of malnutrition in children with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2001;33(3):319-25.
5. Steinkamp G, Wiedemann B. Relationship between nutritional status and lung function in cystic fibrosis: cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax. 2002;57(7):596-601.
6. Pedreira CC, Robert RG, Dalton V, Oliver MR, Carlin JB, Robinson P, et al. Association of body composition and lung function in children with cystic fibrosis. Pediatr Pulmonol. 2005;39(3):276-80.
7. Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2002;35(3):246-59.
8. Thomson MA, Quirk P, Swanson CE, Thomas BJ, Holt TL, Francis PJ, et al. Nutritional growth retardation is associated with defective lung growth in cystic fibrosis: a preventable determinant of progressive pulmonary dysfunction. Nutrition. 1995;11(4):350-4.
9. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998;132(4):589-95.
10. Frisancho AR. New norms of upper limb fat and muscle areas for assessment of nutritional status. Am J Clin Nutr. 1981;34(11):2540-5.
11. Centers for Disease Control and Prevention [homepage on the Internet]. Atlanta: National Center for Health Statistics; c2000-2004 [updated 2001 Apr 20; cited 2004 Feb 10]. 2000 CDC Growth Charts: United States; [about 2 screens]. Available from: http://www.cdc.gov/growthcharts.
12. Tanner JM. Growth at adolescence. Oxford: Blackwell Scientific Publications; 1962.
13. Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948-68.
14. Pencharz PB, Durie PR. Pathogenesis of malnutrition in cystic fibrosis, and its treatment. Clin Nutr. 2000;19(6):387-94.
15. Zemel BS, Jawad AF, FitzSimmons S, Stallings VA. Longitudinal relationship among growth, nutritional status, and pulmonary function in children with cystic fibrosis: analysis of the Cystic Fibrosis Foundation National CF Patient Registry. J Pediatr. 2000;137(3):374-80.
16. Adde FV, Rodrigues JC, Cardoso AL. Nutritional follow-up of cystic fibrosis patients: the role of nutrition education. J Pediatr (Rio J). 2004;80(6):475-82.
17. Lai HC, Kosorok MR, Sondel SA, Chen ST, FitzSimmons SC, Green CG, et al. Growth status in children with cystic fibrosis based on the National Cystic Fibrosis Patient Registry data: evaluation of various criteria used to identify malnutrition. J Pediatr. 1998;132(3 Pt 1):478-85.
18. Dornelas EC, Fernandes MI, Galvão LC, Silva GA. Estudo do quadro pulmonar de pacientes com fibrose cística. J Pediatr (Rio J). 2000;76(4):295-9.
19. Cystic Fibrosis Foundation. Patient Registry 2002 Annual Data Report. Bethesda (MD): Cystic Fibrosis Foundation; 2003.
20. Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, et al. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr. 2003;142(6):624-30.
21. Wiedemann B, Paul KD, Stern M, Wagner TO, Hirche TO; German CFQA Group. Evaluation of body mass index percentiles for assessment of malnutrition in children with cystic fibrosis. Eur J Clin Nutr. 2007;61(6):759-68.
22. Zhang Z, Lai HJ. Comparison of the use of body mass index percentiles and percentage of ideal body weight to screen for malnutrition in children with cystic fibrosis. Am J Clin Nutr. 2004;80(4):982-91.
23. Schöni MH, Casaulta-Aebischer C. Nutrition and lung function in cystic fibrosis patients: review. Clin Nutr. 2000;19(2):79-85.
Study carried out at the Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz, Fernandes Figueira Institute/Oswaldo Cruz Foundation - Rio de Janeiro, Brazil.
Correspondence to: Célia Regina Murtinho de Miranda Chaves. Av. Rui Barbosa, 716, Flamengo, CEP 22250-020, Rio de Janeiro, RJ, Brasil.
Tel 55 21 2554-1718. E-mail: crchaves@iff.fiocruz.br
Financial support: None.
Submitted: 25 August 2008. Accepted, after review: 19 November 2008.
About the authors
Célia Regina Moutinho de Miranda Chaves
Head of the Department of Food and Nutrition. Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz, Fernandes Figueira Institute/Oswaldo Cruz Foundation - Rio de Janeiro, Brazil.
José Augusto Alves de Britto
Pediatrician. Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz, Fernandes Figueira Institute/Oswaldo Cruz Foundation - Rio de Janeiro, Brazil.
Cristiano Queiroz de Oliveira
Physical Trainer. Department of Food and Nutrition, Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz, Fernandes Figueira Institute/Oswaldo Cruz Foundation - Rio de Janeiro, Brazil.
Miriam Martins Gomes
Nutritionist. Department of Food and Nutrition, Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz, Fernandes Figueira Institute/Oswaldo Cruz Foundation - Rio de Janeiro, Brazil.
Ana Lúcia Pereira da Cunha
Nutritionist. Department of Food and Nutrition, Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz, Fernandes Figueira Institute/Oswaldo Cruz Foundation - Rio de Janeiro, Brazil.











 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket