ABSTRACT
Objective: Various studies of emphysema involve long-term exposure of animals to cigarette smoke, focusing on the cell type involved in
the protease/antiprotease imbalance and on extracellular matrix degradation. In emphysema, increased expression of metalloproteinases is
associated with cytokines, and evidence suggests that the matrix metalloproteinase-12 (MMP-12) plays an important role. Our objective was
to investigate tissue inhibitor of metalloproteinase-2 (TIMP-2), tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) detection by
immunohistochemical methods in mouse lung. Methods: Male C57BL/6 mice were exposed 3 times a day to the smoke of 3 cigarettes over
a period of 10, 20, 30 or 60 days in an inhalation chamber (groups CS10, CS20, CS30 and CS60, respectively). Controls were exposed to the
same conditions in room air. Results: A progressive increase in the number of alveolar macrophages was observed in the bronchoalveolar
lavage fluid of the exposed mice. The mean linear intercept, an indicator of alveolar destruction, was greater in all exposed groups when
compared to control group. In the CS10, CS20 and CS30 mice, the immunohistochemical index (II) for MMP-12 increased in parallel with
a decrease in II for TIMP-2 in the CS10, CS20 and CS30 mice. The II for the cytokines TNF-α and IL-6 was greater in all exposed groups
than in the control group. Emphysema, with changes in volume density of collagen and elastic fibers, was observed in the CS60 group.
Conclusions:
These findings suggest that cigarette smoke induces emphysema with major participation of TNF-α and IL-6 without participation
of neutrophils.
Keywords:
Macrophages, alveolar; Emphysema; Tissue inhibitor of metalloproteinase-2; Interleukin-6.
RESUMO
Objetivo: Muitos estudos sobre enfisema são realizados com exposição de animais à fumaça de cigarro durante um longo tempo, focando
o tipo de célula envolvida no desequilíbrio protease/antiprotease e a degradação da matriz extracelular. A expressão aumentada de metaloproteinases
no enfisema está associado com citocinas e evidências sugerem um papel importante da metaloproteinase de matriz-12
(MMP-12). Nosso objetivo foi estudar a detecção de inibidor tissular de metaloproteinase-2 (TIMP-2), fator de necrose tumoral alfa (TNF-α)
e interleucina-6 (IL-6) por métodos imunohistoquímicos no pulmão de camundongos. Métodos: Camundongos C57BL/6 machos foram
expostos 3 vezes ao dia a fumaça de 3 cigarros por um período de 10, 20, 30 ou 60 dias através de uma câmara de inalação (grupos CS10,
CS20, CS30 e CS60, respectivamente). O grupo controle foi exposto às mesmas condições ao ar ambiente. Resultados: Nós observamos um
aumento progressivo de macrófagos alveolares no lavado broncoalveolar dos grupos expostos. O diâmetro alveolar médio, um indicador de
destruição alveolar, aumentou em todos os grupos expostos quando comparado ao grupo controle. O índice imunohistoquímico (II) para
MMP-12 aumentou nos grupos CS10, CS20 e CS30 em paralelo a uma redução do II para TIMP-2 nos grupos CS10, CS20 e CS30. O II para
as citocinas TNF-α e IL-6 aumentou em todos os grupos expostos quando comparado ao grupo controle. Enfisema foi observado no grupo
CS60, com alterações na densidade de volume de fibras colágenas e elásticas. Conclusões: Estes achados sugerem que a fumaça de cigarro
induz enfisema com uma participação importante do TNF-α e da IL-6 sem a participação de neutrófilos.
Palavras-chave:
Macrófagos alveolares; Enfisema; Inibidor tissular de metaloproteinase-2; Interleucina-6.
IntroductionCigarette smoke is a complex mixture with over 4,000 chemical components, being the major risk factor for chronic obstructive pulmonary disease (COPD) and emphysema, the latter appearing to be the more severe consequence.(1) The protease/antiprotease theory in emphysema induced by cigarette smoke is based on a chronic inflammatory response in the lower respiratory tract, where the inflammatory cells liberate a great variety of proteases, which superimposes on the antiproteolytic defenses, with subsequent degradation of the connective tissue.(2) The loss of lung elastic recoil and the histological evidence of damage of the elastic fibers (eFs) necessarily imply degradation of elastin as a key factor in the pathogenesis of the emphysema.(3) Some researchers consider neutrophils to be the principal cells involved in elastolytic injury.(4,5) However, there is evidence that alveolar macrophages are the principal cells responsible for the destruction of the lung architecture.(6,7)
The matrix metalloproteinases (MMPs) participate in the remodeling and degradation of the extracellular matrix.(8) In physiological conditions, the activity of the MMPS are dependent of the activation of zymogens on pro-MMPs and regulated by endogenous inhibitors of MMPs, denominated tissue inhibitors of metalloproteinases (TIMPs).(9) Four types are known: TIMP-1, TIMP-2, TIMP-3 and TIMP-4. All four types can inhibit MMPs with a 1:1 stoichiometry. These are frequently co-expressed with the MMPs, thus controlling the proteolytic effect on the tissue.(10)
Interleukin (IL)-6 is a multifunctional cytokine produced by a variety of cells, generally at sites of inflammation.(11) It acts in different types of cells and systems; however, the role it plays in the pathophysiology of inflammation remains unclear. Recent studies have indicated that IL-6 has a potent anti-inflammatory effect, including the ability to inhibit the expression of tumor necrosis factor alpha (TNF-α), IL-1 and macrophage inflammatory protein 2 (MIP-2), as well as to decrease neutrophil sequestration, increase levels of the IL-1β receptor antagonist, increase the production of TIMPs, increase the intracellular production of superoxide dismutase and inhibit cell apoptosis.(12) Although many cells can secrete TNF-α, macrophages seem to be the major source of its production. The liberation of TNF-α by monocytes or macrophages is markedly increased by other cytokines such as IL-1, granulocyte-macrophage colony-stimulating factor and interferon gamma.(13) Normally synthesized as a 26-kDa precursor, pro-TNF-α is stored in the membrane to be readily liberated, if necessary. When an appropriate stimulus occurs, for example, the inhalation of lipopolysaccharide, TNF-α is rapidly converted into a biologically active, 17-kDa form through the effect of a metalloproteinase known as TNF-α converting enzyme.(14) However, other MMPs also present some degree of TNF-α converting capacity. One such MMP is MMP-12, which presents TNF-α converting capacity, at least in vitro.(15)
Previous studies have shown that TNF-α and MMP-12 play roles in mice exposed to cigarette smoke.(7,16-18) The objective of the present study was to use using immunohistochemical methods in order to investigate the expression of MMP-12, TIMP-2, TNF-α and IL-6 in mice exposed to cigarette smoke.
MethodsMale C57BL/6 mice (6 to 8 weeks of age; weighing 20-25 g) were obtained from the Laboratory Animal Facilities of the Fluminense Federal University, located in the city of Niterói, Brazil. The mice were randomly selected; the health of the animals in the Laboratory Animal Facilities is monitored through laboratory tests for identifying microorganisms. Following quarantine (seven days) the animals were housed in polypropylene cages and maintained on a 12/12-h light/dark cycle with ad libitum access to water and standard chow. The animals were handled according to the standards established by the Roberto Alcântara Gomes Institute of Biology, and the research project was approved by the Ethics Committee for Laboratory Animal Research of that institution.
Groups of male C57BL/6 mice (n = 8 per group) were exposed to the smoke of 3 commercial cigarettes (10 mg of tar, 0.8 mg of nicotine and 10 mg of carbon monoxide), 3 times/day (9 cigarettes/day in total), 7 days/week for 10, 20, 30 or 60 days (CS10, CS20, CS30 and CS60 groups, respectively) in an inhalation chamber as described in previous studies conducted by our group.(7,16-18) In brief, animals in each study group were placed in the inhalation chamber (40 cm in length, 30 cm in width and 25 cm in height). The smoke was produced from each cigarette for 6 minutes and introduced into the exposure chamber using a 60-mL syringe. Each cigarette produced 300 mg/m2 of suspended particulate matter in the chamber. Following exposure to one cigarette, the mice were exposed to 1 min of room air, and the cigarette smoke procedure was repeated. The entire procedure was carried out under a laminar flow hood. Mice in a control group were exposed to the same conditions in room air. One day following the final exposure, the tracheas of 3 mice from each group were cannulated, and the lungs were washed thrice with 0.6 mL of cold phosphate-buffered saline (PBS). Mean volume recovery was 1.5 mL. The total number of cells was determined using a Neubauer chamber. The bronchoalveolar lavage fluid was cytocentrifuged, fixed and stained with Giemsa. The different types of cells were identified through differential counts under light microscopy. Two investigators, blinded to the origin of each slide, conducted the counts.
One day after the final exposure, 5 mice were sacrificed and the lungs were perforated for blood collection through infusion of saline into the right ventricle.
The lungs were fixed in situ for 2 min with buffered formalin (4%) via endotracheal instillation at a pressure of 25 cmH2O. The trachea was blocked, after which the lungs were removed and immersed in fixing solution for 48 h.
After the final paraffin-embedding, 5-µm sections were cut and the slides were stained with hematoxylin-eosin, as is the standard procedure. Picrosirius was used in order to reveal collagen fibers (cFs), and Weigert's resorcin-fuchsin was used in order to reveal eFs. Pulmonary emphysema was quantified based on the degree of alveolar destruction, as determined by measuring the mean linear intercept (Lm, mean alveolar diameter) in micrometers, as previously described.(19) In brief, this technique consists of the determination of the number of times the gas exchange structures in the parenchyma intersect a series of grid lines. Therefore, in the presence of emphysema, the number of intercepts of the alveolar structures with a system of lines will be fewer, indicating alveolar destruction. The Lm is obtained using the equation Lm = Ltot/Li, where Ltot is the total length of the lines in the microscopic field, and Li is the number of intercepts of alveolar structures with the lines of the reticulum. To that end, 16 fields of each slide were counted and observed at a magnification of ×200 through a reticulum attached to the monitor. Stereology was carried out using a test-system composed of 21 points and in a known test area delineated by the forbidden line, in order to avoid overestimation of the number of structures. The test system was attached to a monitor connected to a microscope. The points (PP) that intercepted the cFs and the eFs were counted and compared with the total number of points of a test system (TP). Therefore, the volume density (Vv) is equal to the PP divided by the TP.(17) For the immunohistochemical procedure, the 5-µm sections were incubated with antibodies against MMP-12, TIMP-2, TNF-α and IL-6 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). The antibodies employed, their origins and the protocols used are shown in Table 1. An equal number of slides were submitted to the same procedures without the primary antibodies (negative controls). The immunohistochemical analysis was carried out using the peroxidase-conjugated avidin-biotin complex method (Goat ABC staining system, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), as described in a previous study conducted by our group.(16) In brief, the slides were deparaffinized and rehydrated in a graded xylene-alcohol series. Antigen retrieval was performed using a solution of 1 mM EDTA in citrate buffer (pH 8.0) at high temperature in microwave oven for 5 min. Endogenous peroxidase was inhibited using 3% hydrogen peroxide in methanol. The nonspecific antigen-antibody reaction was blocked through the incubation of the sections in horse serum-PBS and 5-10% horse-serum albumin (HSA)-for 60 min. All antibodies of the negative controls were diluted in PBS/1% HSA for 60 min. Subsequently, all sections were treated with the commercial kit of the secondary antibody (Goat ABC staining system) for 30 min, followed by the peroxidase-conjugated avidin-biotin complex for an additional 30 min. The antigen-antibody reaction was visualized as a brown precipitate following the application of 3,3-diaminobenzidine for 4 min and counterstaining with hematoxylin for 30 s.
The slides were then rehydrated, diaphanized and mounted for observation under light microscopy. The alveolar macrophages were quantified based on the immunohistochemical index (II). To that end, the number of alveolar macrophages testing positive for MMP-12, TIMP-2, TNF-α or IL-6 (MA+) was divided by the total number of macrophages (MAT) × 100, that is, II = MA+/MAT × 100. Therefore, 50 fields of 26,000 µm2 (10 fields in 5 different sections from each group) were counted by two investigators at different times. The investigators, who were blinded to the origin of the slides, performed the counts at a magnification of ×400 using an Olympus microscope (model BH2; Tokyo, Japan). The visualization of the alveolar macrophages in some immunostained slides was confirmed by Giemsa staining.(17,18)
Numerical data were calculated as mean ± standard error. For continuous data, the differences among the groups were determined using analysis of variance followed by Tukey's post-test. For categorical data, the differences among the groups were determined using the Kruskal-Wallis test followed by Dunn's post-test. In both cases, values of p < 0.05 were considered significant.
ResultsAs can be seen in Figure 1, the number of alveolar macrophages was significantly higher in the CS10 group than in the control group (p < 0.001), and this difference became greater in direct relation to increases in the duration of exposure (p < 0.001 for the CS20, CS30 and CS60 groups vs. controls).
However, no significant differences in the number of neutrophils were observed. In the histological analysis, samples from the control group animals (exposed to room air only) presented preserved alveoli with thin alveolar septa and a minimal number of alveolar macrophages (Figure 2a). The CS10 and CS20 group samples presented a histological pattern with progressive alterations of breakage of alveolar septa with a greater number of alveolar macrophages. The CS30 group samples presented an initial pattern of alterations in the lung architecture principally with enlargements of the air spaces corresponding to the alveolar duct and alveoli. The CS60 group samples presented a typical pattern of murine emphysema, with alveolar enlargement and areas of initial fibrosis (Figure 2b).
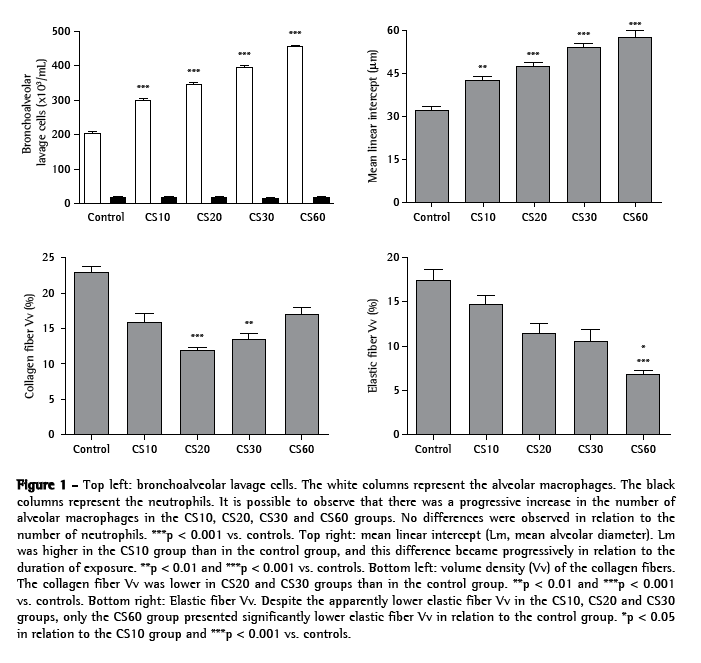
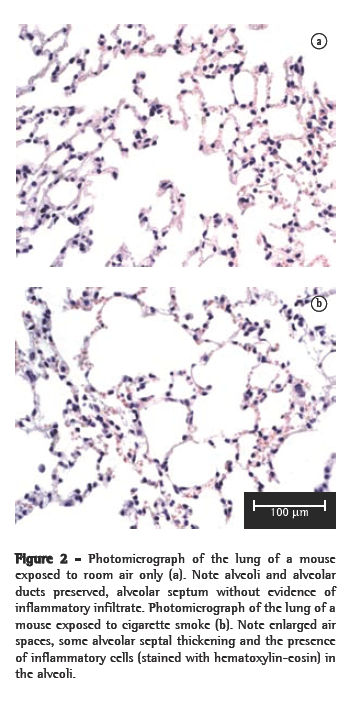
The alterations in the lung architecture were quantified based on the Lm, Vv(cF) and Vv(eF), as is also shown in Figure 1. In comparison with the control group values, Lm was significantly greater in the CS10 group (p < 0.01), and this difference became progressively larger in the CS20, CS30 and CS60 groups (p < 0.001). Similarly, Vv(cF) was significant lower in the CS20 (p < 0.001) and CS30 (p < 0.01) groups than in the control group. In addition, Vv(eF) was greater in the control group than in the CS10, CS20 and CS30 groups, although the difference was not statistically significant. In the CS60 group, Vv(eF) was significantly lower than in the control group (p < 0.001) and in the CS10 group (p < 0.05).
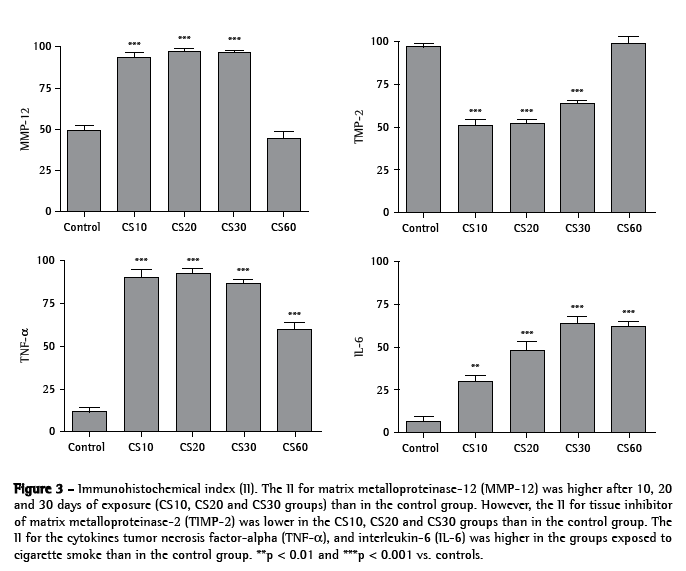
Levels of MMP-12, TIMP-2, TNF-α and IL-6 were quantified based on the II. All alveolar macrophages, positive or not, were counted. As shown in Figures 3, 4a, 4b and 4c, the II for MMP-12 was higher in the CS10, CS20 and CS30 groups than in the control group (p < 0.001). Figures 3, 4d, 4e and 4f show that the II for TIMP-2 was lower in the CS10, CS20 and CS30 groups than in the control group (p < 0.001). All CS groups presented a higher II for TNF-α than did the control group (p < 0.001; Figures 3, 4g, 4h and 4i). However, the II for TNF-α was lower in the CS60 group than in the CS10, CS20 and CS30 groups. As shown in Figures 3, 4j, 4l and 4m, the II for IL-6 was progressively higher, in comparison with the control group, in the CS10, CS20, CS30 and CS60 groups (p < 0.01, p < 0.001, p < 0.001 and p < 0.001, respectively).
 Discussion
DiscussionAirflow obstruction due to bronchitis or emphysema are the defining features of COPD.(20) A variety of MMPs can participate in COPD through the degradation of components of the extracellular matrix, principally elastin.(7,17,21) However, MMPs also play a role in regulating inflammation through the generation of chemokines and cytokines and by creating openings in the tissue barriers for the passage of inflammatory cells.(22) Emphysema probably occurs when the cigarette smoke causes extensive and continuous recruitment of inflammatory cells, which liberate proteolytic enzymes into the extracellular matrix. The destruction of the lung parenchyma is permanent due to the failure to repair the enlargement of the air spaces, which are characteristic of emphysema.(23)
In a previous study, septal thickening and air space enlargement were described in C57BL/6 mice after 60 days of exposure to cigarette smoke.(7)
These alterations were followed by lower numbers of cFs and eFs parallel to an increase in the number of alveolar macrophages. The present study was carried out in order to gain a better understanding of the participation of alveolar macrophages in this model of emphysema. Here, emphysema was identified based on an increase in the Lm, an index widely used for the quantification of the alveolar destruction.(19) Despite the significant increase in Lm observed in the CS10 group, the histological pattern of emphysema was evident only in the CS60 group. The imbalance between protease and antiprotease, evidenced here by the immunohistochemical detection of MMP-12 and TIMP-2, was also indicated by the presence of TNF-α and IL-6. These results indicated an increase in MMP-12 concomitant to a decrease in TIMP-2 in the CS10, CS20 and CS30 groups. However, in the group that presented a histological pattern of emphysema (CS60), it seems that MMP-12 did not play such an important role, which, it is believed, is partially, due to the increase in TIMP-2. However, Vv(eF) was progressively reduced according to the time of exposure, the maximum reduction (in the CS60 group) being significant in relation to that observed for the control group. Curiously, the significant reduction in the number of eFs occurred simultaneous to the decrease in the expression of MMP-12 (after 60 days of exposure). It is known that MMP-12 plays a role in emphysema, in mice,(7,17,24) as well as in humans.(6) However, little is known about the role played by TIMP-2. In the present study, we suggest that, after the disease is established, the production of MMP-12 becomes less important. Therefore, the increased production of TIMP-2 in the CS60 group might be responsible for the increase in Vv(cF), as determined using stereology. Other proteolytic enzymes can participate in the injurious process that leads to emphysema, as in the case of gelatinases, collagenases and stromelysins.(21) It is impossible to state-nor is it the objective of the present study to state-that the antiprotease effects are attributable to TIMP-2 alone, since other TIMPs have been shown to have some effect in emphysema, in animal models,(9) as well as in humans.(21)
In the present study, levels of TNF-α and IL-6 in alveolar macrophages were also analyzed. In the control group, the II for TNF-α and IL-6 was low. In the CS10, CS20, CS30 and CS60 groups, the II for TNF-α was significantly higher than in the control group. This finding corroborates those of previous studies,(12,16,17,25) which showed a special participation of TNF-α in emphysema induced by cigarette smoke. The II for IL-6 increased progressively in direct relation to increases in the duration of exposure. The present study is the first to show that IL-6 plays a significant role in pulmonary inflammation (after 10 and 20 days of exposure) and emphysema (after 60 days of exposure). Our results indicate that IL-6 plays a pro-inflammatory role and can somehow be involved in COPD. The concentration of IL-6 in the bronchoalveolar lavage has been shown to be significantly higher in patients with COPD than in those without, suggesting that smoking plays some role in the increased concentration of IL-6.(12) In addition, IL-6 knockout mice present reduced liberation of cytokines, chemokines and other pro-inflammatory mediators.(26,27)
The results suggest that the secretion, liberation and activity of MMP-12, as observed here, allow the liberation of TNF-α. This relationship has previously been described.(25) We also suggest that the presence of TNF-α is not exclusive to an acute phase or related only to the presence of neutrophils. However, it is important to remember that, in the present study, we did not analyze the inflammatory cells of the alveolar septum, which we believe to be as important for the course of the COPD as are the inflammatory cells present in the alveoli. Although the protease/antiprotease theory is widely accepted, the cells and the types of proteolytic enzymes that certainly participate in the pathological process constitute a controversial matter. Nor is the oxidant/antioxidant theory to be ignored.(28) In that theory, oxidative stress is considered a major contributor to the pathogenesis and progression of COPD. Reactive oxygen and nitrogen species in cigarette smoke and in the inflammatory cells react with the extracellular matrix, thereby inducing tissue damage.(29) Although inflammatory mediators also increase the production of oxidants in the inflammatory cells,(28,29) it is unclear whether the exclusion of the inflammatory mediators could allow the occurrence of lung injury induced exclusively by oxidants.
In conclusion, we suggest that the macrophage is a key cell in the destruction of the extracellular matrix in the lung, the temporal progression of which leads to emphysema in mice. As shown in other studies, MMP-12 appears to play an important role in the onset of emphysema, and production of MMP-12 can be reduced by an increase in that of TIMP-2, albeit without any influence on the final course of the pathological process. As markers of emphysema, TNF-α and IL-6 are both important, and our findings suggest that both have a pro-inflammatory effect.
References 1. Borgerding M, Klus H. Analysis of complex mixtures--cigarette smoke. Exp Toxicol Pathol. 2005;57(Suppl1):43-73.
2. Hoyt JC, Robbins RA, Habib M, Springall DR, Buttery LD, Polak JM, et al. Cigarette smoke decreases inducible nitric oxide synthase in lung epithelial cells. Exp Lung Res. 2003;29(1):17-28.
3. Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56(4):515-48.
4. Dhami R, Gilks B, Xie C, Zay K, Wright JL, Churg A. Acute cigarette smoke-induced connective tissue breakdown is mediated by neutrophils and prevented by alpha1-antitrypsin. Am J Respir Cell Mol Biol. 2000;22(2):244-52.
5. Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163(6):2329-35.
6. Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res. 2005;54(1):31-6.
7. Valença SS, da Hora K, Castro P, Moraes VG, Carvalho L, Porto LC. Emphysema and metalloelastase expression in mouse lung induced by cigarette smoke. Toxicol Pathol. 2004;32(3):351-6.
8. Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006;11:1696-701.
9. Bode W, Maskos K. Structural studies on MMPs and TIMPs. Methods Mol Biol. 2001;151:45-77.
10. Gueders MM, Foidart JM, Noel A, Cataldo DD. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: potential implications in asthma and other lung diseases. Eur J Pharmacol. 2006;533(1-3):133-44.
11. Chyczewska E, Mróz RM, Kowal E. TNF-alpha, IL-1 and IL-6 concentration in bronchoalveolar lavage fluid (BALF) of non-small cell lung cancer (NSCLC). Rocz Akad Med Bialymst. 1997;42(Suppl 1):123-35.
12. Seifart C, Dempfle A, Plagens A, Seifart U, Clostermann U, Müller B, et al. TNF-alpha-, TNF-beta-, IL-6-, and IL-10-promoter polymorphisms in patients with chronic obstructive pulmonary disease. Tissue Antigens. 2005;65(1):93-100.
13. Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2001;34:S50-S59.
14. Majka S, McGuire PG, Das A. Regulation of matrix metalloproteinase expression by tumor necrosis factor in a murine model of retinal neovascularization. Invest Ophthalmol Vis Sci. 2002;43(1):260-6.
15. Gilks CB, Price K, Wright JL, Churg A. Antioxidant gene expression in rat lung after exposure to cigarette smoke. Am J Pathol. 1998;152(1):269-78.
16. da Hora K, Valença SS, Porto LC. Immunohistochemical study of tumor necrosis factor-alpha, matrix metalloproteinase-12, and tissue inhibitor of metalloproteinase-2 on alveolar macrophages of BALB/c mice exposed to short-term cigarette smoke. Exp Lung Res. 2005;31(8):759-70.
17. Valenca SS, Castro P, Pimenta WA, Lanzetti M, Silva SV, Barja-Fidalgo C, et al. Light cigarette smoke-induced emphysema and NFkappaB activation in mouse lung. Int J Exp Pathol. 2006;87(5):373-81.
18. Silva Bezerra F, Valença SS, Lanzetti M, Pimenta WA, Castro P, Gonçalves Koatz VL, et al. Alpha-tocopherol and ascorbic acid supplementation reduced acute lung inflammatory response by cigarette smoke in mouse. Nutrition. 2006;22(11-12):1192-201.
19. Fusco LB, Pêgo-Fernandes PM, Xavier AM, Pazetti R, Rivero DH, Capelozzi VL, et al. Modelo experimental de enfisema pulmonar em ratos induzido por papaína. J Pneumol. 2002;28(1):1-7.
20. Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343(4):269-80.
21. Belvisi MG, Bottomley KM. The role of matrix metalloproteinases (MMPs) in the pathophysiology of chronic obstructive pulmonary disease (COPD): a therapeutic role for inhibitors of MMPs? Inflamm Res. 2003;52(3):95-100.
22. Shapiro SD. Proteinases in chronic obstructive pulmonary disease. Biochem Soc Trans. 2002;30(2):98-102.
23. Lagente V, Manoury B, Nénan S, Le Quément C, Martin-Chouly C, Boichot E. Role of matrix metalloproteinases in the development of airway inflammation and remodeling. Braz J Med Biol Res. 2005;38(10):1521-30.
24. Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002-4.
25. Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170(5):492-8.
26. Cataldo D, Munaut C, Noël A, Frankenne F, Bartsch P, Foidart JM, et al. MMP-2- and MMP-9-linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol. 2000;123(3):259-67.
27. Kuhn C 3rd, Homer RJ, Zhu Z, Ward N, Flavell RA, Geba GP, et al. Airway hyperresponsiveness and airway obstruction in transgenic mice. Morphologic correlates in mice overexpressing interleukin (IL)-11 and IL-6 in the lung. Am J Respir Cell Mol Biol. 2000;22(3):289-95.
28. Rahman I, Biswas SK, Kode A. Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol. 2006;533(1-3):222-39.
29. Domej W, Földes-Papp Z, Flögel E, Haditsch B. Chronic obstructive pulmonary disease and oxidative stress. Curr Pharm Biotechnol. 2006;7(2):117-23.
Trabalho realizado na Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.
1. Professora da Pontifícia Universidade Católica de São Paulo, São Paulo (SP) Brasil.
2. Pos-Doutorando na Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.
3. Professor no Departamento de Histopatologia do Royal Brompton Hospital, London, Reino Unido.
4. Professor de Matemática e Estatística da Universidade de São Paulo, São Paulo (SP) Brazil.
5. Professor Associado do Departamento de Patologia da Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.
Endereço para correspondência: Vera Luiza Capelozzi or Edwin Roger Parra, Departamento de Patologia Faculdade de Medicina da USP, Av. Dr. Arnaldo, 455, Sala 1143, CEP 01246-903, São Paulo (SP) Brazil.
Tel 55 11 3061-7427; Fax 55 11 3064-2744. E-mail: vcapelozzi@lim05.fm.usp.br / erparra20003@yahoo.com.br
Apoio financeiro: Este trabalho recebeu apoio financeiro das seguintes agências brasileiras: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Processo 300430/95-7; Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Processo 2000/14336-0; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); e Laboratórios de Investigação Médica (LIM) do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
Recebido para publicação em 14/11/2007. Aprovado, após revisão, em 30/1/2008.













 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket