ABSTRACT
Objective: To evaluate spirometric patterns of respiratory disorders and their relationship with functional severity and maximal expiratory flows at low lung volumes in patients with cystic fibrosis (CF). Methods: A retrospective cross-sectional study including adolescents and adults with CF. All of the patients were submitted to spirometry. Patients were classified as having preserved respiratory function, obstructive lung disease (OLD), OLD with reduced FVC, presumptive restrictive lung disease (RLD) or mixed obstructive and restrictive lung disease (MORLD). Maximal expiratory flows at low lung volumes were assessed using FEF25-75%, FEF75% and FEF75%/FVC. We included 65 normal subjects, also submitted to spirometry, as a control group. Results: The study group included 65 patients: 8 (12.3%) with preserved lung function; 18 (27.7%) with OLD; 24 (36.9%) with OLD and reduced FVC; 5 (7.7%) with presumptive RLD; and 10 (15.4%) with MORLD. The FEV1 was significantly lower in the OLD with reduced FVC group and the MORLD group than in the other groups (p < 0.001). In the patients with preserved respiratory function, FEF25-75% and FEF75% were significantly reduced in 1 patient, as was FEF75%/FVC in 2 patients. Conclusions: The respiratory pattern was impaired in 88% of the patients with CF. The most common pattern was OLD with reduced FVC. The degree of functional impairment was greater in the OLD with reduced FVC group and in the MORLD group than in the other groups. Maximal expiratory flows at low lung volumes were impaired in a low percentage of patients with preserved respiratory function.
Keywords:
Cystic fibrosis; Respiratory function tests; Spirometry; Maximal expiratory flow-volume curves.
RESUMO
Objetivo: Avaliar os padrões dos distúrbios ventilatórios observados na espirometria em pacientes com fibrose cística (FC) e suas relações com a gravidade funcional e com o comportamento dos fluxos máximos expiratórios a baixos volumes. Métodos: Estudo transversal e retrospectivo, incluindo pacientes adolescentes e adultos com FC. Todos os pacientes foram submetidos à espirometria. Os pacientes foram classificados como tendo função ventilatória preservada, distúrbio ventilatório obstrutivo (DVO), DVO com CVF reduzida, sugestivo de distúrbio ventilatório restritivo (DVR) ou distúrbio ventilatório combinado (DVC). Os fluxos máximos expiratórios a baixos volumes foram avaliados utilizando-se FEF25-75%, FEF75% e FEF75%/CVF. O grupo controle incluiu 65 indivíduos normais, também submetidos à espirometria. Resultados: Foram incluídos 65 pacientes no grupo de estudo: 8 (12,3%) com função pulmonar preservada, 18 (27,7%) com DVO, 24 (36,9%) com DVO com CVF reduzida, 5 (7,7%) com padrão sugestivo de DVR e 10 (15,4%) com DVC. O VEF1 foi significativamente menor nos grupos DVO com CVF reduzida e DVC, comparados com os outros grupos (p < 0,001). Nos pacientes com função ventilatória preservada, FEF25-75% e FEF75% foram significativamente reduzidos em 1 paciente, assim como FEF75%/CVF em 2 pacientes. Conclusões: O padrão ventilatório estava alterado em 88% dos pacientes com FC. O distúrbio mais frequente foi DVO com CVF reduzida. Houve maior prejuízo funcional nos pacientes com DVO e CVF reduzida e com DVC. Os fluxos expiratórios máximos a baixos volumes foram alterados em uma pequena percentagem de pacientes com função pulmonar preservada.
Palavras-chave:
Fibrose cística; Testes de função respiratória; Espirometria; Curvas de fluxo-volume máximo expiratório.
IntroductionCystic fibrosis (CF), or mucoviscidosis, is a lethal, autosomal recessive genetic disease that is more common in Whites. It results from exocrine cell dysfunction, which is responsible for the multisystemic involvement in CF.(1)
Since CF is characterized by a wide phenotypic variability, there are significant clinical differences among patients in terms of disease severity and complications. The major clinical repercussions are related to pulmonary involvement, and respiratory symptoms account for 90% of cases of morbidity and mortality.(2)
In the lungs, abnormalities in ionic transport promote a reduction in mucosal secretions, an increase in viscosity of these secretions and a consequent decrease in mucociliary clearance. At birth, the lungs are practically normal, although there is already mucoid impaction in the pulmonary mucous glands. The retention of thick mucous in the bronchioles favors the onset of a vicious cycle of inflammation, bacterial infection, destruction of bronchial architecture and bronchiectasis.(3-5)
Survival in CF has increased significantly in the past 30 years due to therapeutic advances and the consequent delay in the progressive decline in lung function. Periodic assessment of lung function in patients with CF and early detection of airway alterations play an important role in treatment, contributing to a decrease in morbidity and mortality rates.(6)
Spirometry is regularly used in the assessment and follow-up of patients with CF. In general, spirometry is performed in each outpatient visit, making it possible to identify the presence of a respiratory disorder and to determine the degree of airway involvement. It is also useful in monitoring the response to therapeutic interventions. Assessment and monitoring of end-expiratory flow parameters would allow the prevention and early detection of major functional changes.(7-9)
The objective of the present study was to assess spirometric patterns of respiratory disorders and determine their relationship with functional severity and end-expiratory flow parameters in adolescents and adults with CF treated at a referral center for the disease.
MethodsThis was a retrospective cross-sectional study carried out at the Hospital de Clínicas de Porto Alegre (HCPA, Porto Alegre Hospital de Clínicas). The study design was approved by the HCPA Research and Postgraduate Group Ethics Committee (Protocol no. 06060), and all participants gave written informed consent.
The medical charts of all of the 65 patients treated via the HCPA Program for Adolescents and Adults with CF were reviewed. We included patients with a diagnosis of CF, confirmed in accordance with the criteria established in the Cystic Fibrosis Foundation Consensus statement,(10) who were treated via the HCPA Program for Adolescents and Adults with CF.
We selected a control group of healthy patients with normal lung function, as defined by the criteria established in the Brazilian Guidelines for Pulmonary Function Tests. Those individuals were selected from the database of the HCPA pulmonary function laboratory. The control subjects were matched to the study subjects in terms of gender and age.
The demographic characteristics of the patients with CF were obtained from the database of a previous study involving patients treated via the HCPA Program for Adolescents and Adults with CF.
The weight and height of patients were recorded, and body mass index (BMI) was calculated as the weight in kilograms divided by the square of the height in meters.
All spirometric tests used in the present study were performed in the HCPA Pulmonary Physiology Unit. Pulmonary function tests were performed using a computerized spirometer (Jaeger - v 4.31; Würzburg; Germany). Maximal vital capacity (MVC), FVC and FEV1 were recorded, as were the FEV1/MVC ratio, the FEV1/FCV ratio, mean forced expiratory flow between 25 and 75% of forced vital capacity (MFEF25-75%), forced expiratory flow at 75% of forced vital capacity (FEF75%) and the FEF75%/FVC ratio. During the spirometric test, three forced expiratory maneuvers were performed, and the best result was registered. All values were expressed in liters and in percentage of the predicted values for age, height and gender.(11) Spirograms were interpreted and respiratory disorders were characterized in accordance with the 2002 Brazilian Guidelines for Pulmonary Function Tests.(12)
For the statistical analysis, the patients with CF were classified in two ways: by type of respiratory disorder-normal spirometry results (N), obstructive lung disease (OLD), OLD with reduced FVC, presumptive restrictive lung disease (RLD) and mixed obstructive and restrictive lung disease (MORLD); and by severity of the respiratory disorder-normal spirometry results (FEV1 ≥ 80% of predicted), mild respiratory disorder (FEV1 ≥ 40-60% of predicted) and severe respiratory disorder (FEV1 < 40% of predicted).
Data were expressed as number of cases (proportion) and as mean ± SD. One-way ANOVA, followed by Tukey's post-hoc test, was used to compare continuous variables among three or more groups. Data were analyzed using the program Statistical Package for the Social Sciences, version 13.0 (SPSS Inc., Chicago, IL, USA). The level of statistical significance was set at p < 0.05. All statistical tests were two-tailed.
ResultsAll of the 65 patients treated via the HCPA Program for Adolescents and Adults with CF were included in the study. The mean age of the patients was 24.5 ± 7.2 years (range 14-49 years). The mean BMI was 21.0 ± 2.5 kg/m2 (range: 16-27 kg/m2). Of the 65 patients, 37 were female and 28 were male. The mean FEV1 and the mean FVC were 57.2 ± 26.2% of predicted and 71.6 ± 22.9% of predicted, respectively (Table 1).
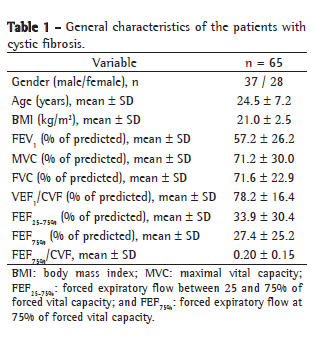
Table 2 compares the spirometric variables by respiratory disorder. In our sample, 8 patients (12.3%) were classified as presenting normal spirometry results, 18 (27.7%) were classified as having OLD, 24 (36.9%) were classified as having OLD with reduced FVC, 10 (15.4%) were classified as having MORLD and 5 (7.7%) were classified as having presumptive RLD. The FEV1 values were significantly lower in the OLD with reduced FVC group and in the MORLD group than in the other groups-the OLD group, the presumptive RLD group and the preserved lung function group (p < 0.001). Of the 8 patients classified as having preserved lung function, only 1 presented MFEF25-75% and FEF75% values below the lower limit of normality and 2 presented values FEF75%/FVC below 0.25.
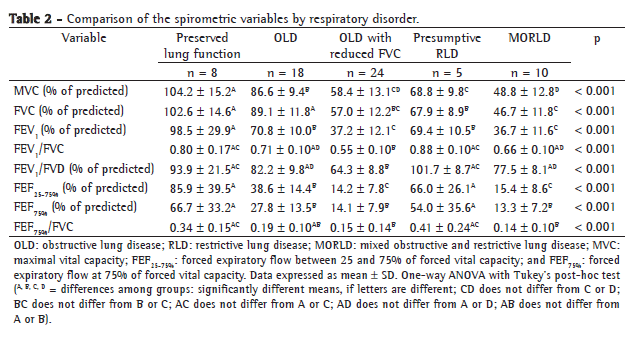
Table 3 compares the spirometric variables by functional severity. Of the 65 patients, 9 (13.8%) presented FEV1 ≥ 80%, 18 (27.7%) presented FEV1 ≥ 60-80%, 18 (27.7%) presented FEV1 ≥ 40-60% and 20 presented FEV1 < 40%.
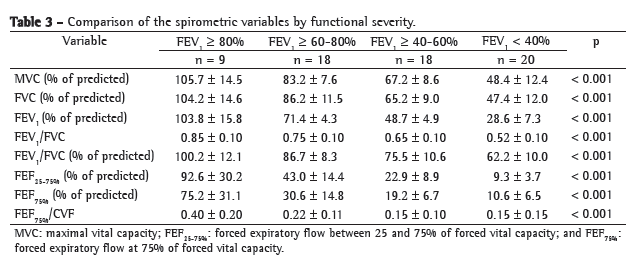
Figure 1 compares the normal individuals and the CF patients, grouped by type of respiratory disorder, in terms of FEF75%/FVC value. There were no statistically significant differences among the control group, the preserved lung function group and the presumptive RLD group, although these groups differed significantly from the OLD group, the OLD with reduced FVC group and the MORLD group, which did not differ among themselves (p < 0.001).
 Discussion
DiscussionIn this cross-sectional study, we observed that the respiratory pattern was impaired in 88% of the adolescents and adults with CF, as demonstrated by spirometry, whereas lung function was preserved in only 12%. The most common pattern of impairment was OLD with reduced FVC (in 37%), followed by OLD (in 28%), MORLD (in 15%) and presumptive RLD (in 8%). There was a statistically significant association between type of respiratory disorder and functional severity as determined by FEV1, OLD with reduced FVC and MORLD being the patterns associated with the greatest functional severity.
The assessment of end-expiratory flow parameters in patients with preserved lung function revealed that 3 patients showed impairment: 1 showed a reduction in FEF25-75% and FEF75%; and 2 showed a reduction in FEF75%/FVC. This might express an early involvement of the small airways caused by the inflammatory process of the disease.
Regarding the assessment of functional severity as determined by FEV1, 37% of the patients had mild lung disease, 28% had moderate lung disease and 31% had severe lung disease.
Abnormalities in respiratory function in patients with CF correlate with the severity of changes in lung structure and with clinical manifestations. In CF, the most common change in pulmonary function is a predominantly obstructive pattern with air trapping, and, in advanced stages of the disease, the onset of a restrictive process due to pulmonary fibrosis.(13,14)
Although CF was once exclusively a childhood disease, increasing knowledge about the disease and the improvements in treatment have allowed an increasingly greater survival, making it possible for a significant number of individuals with CF to reach adolescence and adulthood. However, as these individuals reach puberty and progress into adulthood, there is inherent disease progression and worsening of lung function.(15) In a retrospective cohort study of 52 patients (age ranging from 4 to 26 years; mean, 13.04 ± 4.82 years) treated for approximately 12 years and assessed by spirometry (243 tests) with a bronchodilator test, there was a progressive reduction in the mean of the spirometric parameters studied. At the end of the observation period, all of the patients were found to present reduced respiratory capacity, and OLD predominated. There was an early reduction in end-expiratory flow parameters (FEF50% and FEF25-75%) and late impairment of FVC.(16)
In young individuals without clinical pulmonary manifestations and presenting essentially normal spirometry results, spirometry can demonstrate early involvement of the peripheral airways through the evaluation, in the flow-volume curve, of the instantaneous flows at low lung volumes (the last 25% of FVC).(17) These flows can be adjusted for total FVC(18,19) or for the corresponding segment of FVC (FEF75% at "isovolume").(20)
The principal limitation of the present study was that it employed a cross-sectional design, which does not provide sufficient evidence to establish the temporal sequence between the progression of disease severity and the patterns of alterations, as determined by spirometry.
In conclusion, we observed that, in 88% of the adolescents and adults with CF treated at a referral center for the disease, the respiratory pattern was impaired, as determined by spirometry. The most common pattern of impairment was OLD with reduced FVC, followed by OLD. The patterns associated with greater functional severity were OLD with reduced FVC and MORLD. The analysis of end-expiratory flow parameters allowed us to identify an additional impairment in a small percentage of patients classified as having preserved lung function.
References 1. Bush A, Alton EW, Davies JC, Griesenbach U, Jaffe A. Cystic Fibrosis in the 21st Century. Respir Care. 2006;51(8):930.
2. Ribeiro JD, Ribeiro MA, Ribeiro AF. Controversies in cystic fibrosis--from pediatrician to specialist [Article in Portuguese]. J Pediatr (Rio J). 2002;78 Suppl 2:S171-86.
3. Chmiel JF, Konstan MW. Inflammation and anti-inflammatory therapies for cystic fibrosis. Clin Chest Med. 2007;28(2):331-46.
4. Morrissey BM. Pathogenesis of bronchiectasis. Clin Chest Med. 2007;28(2):289-96.
5. Schidlow DV, Taussig LM, Knowles MR. Cystic Fibrosis Foundation consensus conference report on pulmonary complications of cystic fibrosis. Pediatr Pulmonol. 1993;15(3):187-98.
6. Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125(1 Suppl):1S-39S.
7. Rosenthal M. Annual assessment spirometry, plethysmography, and gas transfer in cystic fibrosis: do they predict death or transplantation. Pediatr Pulmonol. 2008;43(10):945-52.
8. Ren CL, Pasta DJ, Rasouliyan L, Wagener JS, Konstan MW, Morgan WJ, et al. Relationship between inhaled corticosteroid therapy and rate of lung function decline in children with cystic fibrosis. J Pediatr. 2008;153(6):746-51.
9. Konstan MW, Morgan WJ, Butler SM, Pasta DJ, Craib ML, Silva SJ, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr. 2007;151(2):134-9, 139.e1.
10. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998;132(4):589-95.
11. Pereira CA, Sato T, Rodrigues SC. New reference values for forced spirometry in white adults in Brazil. J Bras Pneumol. 2007;33(4):397-406.
12. Sociedade Brasileira de Pneumologia e Tisiologia. Diretrizes para testes de função pulmonar. J Pneumol. 2002;28(3):S1-S238.
13. Zapletal A, Motoyama EK, Gibson LE, Bouhuys A. Pulmonary mechanics in asthma and cystic fibrosis. Pediatrics. 1971;48(1):64-72.
14. Packe GE, Hodson ME. Changes in spirometry during consecutive admissions for infective pulmonary exacerbations in adolescent and adult cystic fibrosis. Respir Med. 1992;86(1):45-8.
15. Dalcin Pde T, Abreu E, Silva FA. Cystic fibrosis in adults: diagnostic and therapeutic aspects. J Bras Pneumol. 2008;34(2):107-17.
16. Andrade EF, Fonseca DL, Abreu e Silva FA, Menna-Barreto SS. Avaliação evolutiva da espirometria na fibrose cística. J Pneumol. 2001;27(3):130-6.
17. Lamarre A, Reilly BJ, Bryan AC, Levison H. Early detection of pulmonary function abnormalities in cystic fibrosis. Pediatrics. 1972;50(2):291-8.
18. Knudson RJ, Burrows B, Lebowitz MD. The maximal expiratory flow-volume curve: its use in the detection of ventilatory abnormalities in a population study. Am Rev Respir Dis. 1976;114(5):871-9.
19. Gilbert R, Auchincloss JH Jr. The interpretation of the spirogram. How accurate is it for 'obstruction'? Arch Intern Med. 1985;145(9):1635-9.
20. Meyer FJ, Ewert R, Hoeper MM, Olschewski H, Behr J, Winkler J, et al. Peripheral airway obstruction in primary pulmonary hypertension. Thorax. 2002;57(6):473-6.
Study carried out in the Department of Pulmonology, Porto Alegre Hospital de Clínicas, Porto Alegre, Brazil.
Correspondence to: Bruna Ziegler. Rua Ramiro Barcelos. 1690/202, Rio Branco, CEP 90.035-002, Porto Alegre, RS, Brasil.
Tel 55 51 3335-1286. E-mail: brunaziegler@yahoo.com.br
Financial support: This study received financial support from the Fundo de Incentivo à Pesquisa (FIPE, Research Incentive Fund) of the Porto Alegre Hospital de Clínicas, Universidade Federal do Rio Grande do Sul (UFRGS, Federal University of Rio Grande do Sul) School of Medicine.
Submitted: 22 January 2009. Accepted, after review: 4 May 2009.
About the authorsBruna Ziegler
Physical Therapist. Department of Pulmonology, Porto Alegre Hospital de Clínicas, Porto Alegre, Brazil.
Paula Maria Eidt Rovedder
Adjunct Professor. School of Physical Therapy, Methodist University Center of the Instituto Porto Alegre - IPA, Porto Alegre Institute - Porto Alegre, Brazil.
Paulo de Tarso Roth Dalcin
Adjunct Professor. Federal University of Rio Grande do Sul School of Medicine, Porto Alegre, Brazil.
Sérgio Saldanha Menna-Barreto
Full Professor. Department of Internal Medicine, Federal University of Rio Grande do Sul School of Medicine, Porto Alegre, Brazil.











 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket