
Continuous and bimonthly publication
ISSN (on-line): 1806-3756



Antonio George de Matos Cavalcante, Pedro Felipe Carvalhedo de Bruin
ABSTRACT
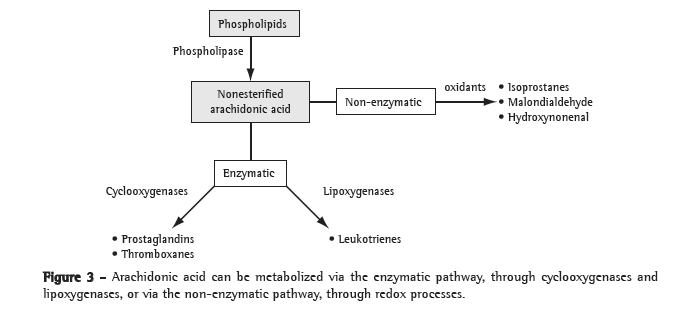
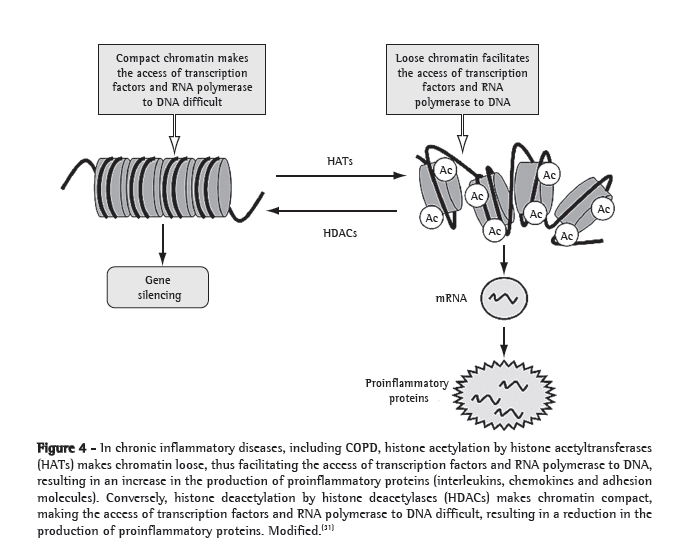
Worldwide, COPD is a major cause of morbidity and mortality. The clinical and functional manifestations of COPD result from lung injury occurring through various mechanisms, including oxidative stress, inflammation, protease-antiprotease imbalance and apoptosis. Oxidative stress is central to the pathogenesis of COPD, since it can directly damage lung structures and exacerbate the other mechanisms involved. The cellular and molecular events involved in such lung injury are believed to occur long before the clinical and functional expression of COPD. Although the use of bronchodilators is currently the principal treatment for COPD, bronchodilators have little or no effect on disease progression. A better understanding of the pathogenesis of COPD, together with renewed efforts in basic and clinical research, will allow the diagnosis of COPD at a pre-clinical stage and provide more appropriate monitoring of disease activity, as well as leading to the development of novel therapeutic agents that will effectively prevent the progression of the disease.
Keywords: Pulmonary disease, chronic obstructive; Oxidative stress; Oxidants; Antioxidants; Inflammation.
RESUMO
A DPOC é uma causa importante de morbidade e mortalidade em escala global. As manifestações clínicas e funcionais da DPOC resultam de danos pulmonares provocados por um conjunto de mecanismos, incluindo o estresse oxidativo, a inflamação, o desequilíbrio do sistema protease-antiprotease e a apoptose. O estresse oxidativo é central na gênese da DPOC, pois além de provocar dano direto às estruturas pulmonares, amplifica os demais mecanismos. Os eventos celulares e moleculares responsáveis pelo dano pulmonar antecedem em muito a expressão clínica e funcional da DPOC. Os broncodilatadores, principais drogas empregadas atualmente no tratamento da DPOC, não são eficazes em reduzir a progressão da doença. Avanços na compreensão da patogênese da DPOC aliados a esforços renovados na pesquisa básica e clínica deverão permitir sua detecção na fase pré-clínica e possibilitar um monitoramento mais adequado de sua atividade, além de permitir a introdução de novas modalidades de agentes terapêuticos capazes de impedir eficazmente sua progressão.
Palavras-chave: Doença pulmonar obstrutiva crônica; Estresse oxidativo; Oxidantes; Antioxidantes; Inflamação.
Introduction








Sociedade Brasileira de Pneumologia e Tisiologia
Secretaria do Jornal Brasileiro de Pneumologia
SCS Quadra 01, Bloco K, Salas 203/204 Ed. Denasa
Brasília - DF CEP: 70.398-900
Fone/fax: 08000 61 6218, (61) 3245 1030 e (61) 3245 6218
E-mail: jbp@sbpt.org.br
Development by: ![]()
© All rights reserved 2025 - Jornal Brasileiro de Pneumologia
 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket