ABSTRACT
Although mechanical ventilation is an important therapy, it can result in complications. One major complication is ventilator-induced lung injury, which is caused by alveolar hyperdistension, leading to an inflammatory process, with neutrophilic infiltration, hyaline membrane formation, fibrogenesis and impaired gas exchange. In this process, cellular mechanotransduction of the overstretching stimulus is mediated by means of the cytoskeleton and its cell-cell and cell-extracellular matrix interactions, in such a way that the mechanical stimulus of ventilation is translated into an intracellular biochemical signal, inducing endothelial activation, pulmonary vascular permeability, leukocyte chemotaxis, cytokine production and, possibly, distal organ failure. Clinical studies have shown the relationship between pulmonary distension and mortality in patients with ventilator-induced lung injury. However, although the cytoskeleton plays a fundamental role in the pathogenesis of ventilator-induced lung injury, there have been few in vivo studies of alterations in the cytoskeleton and in cytoskeleton-associated proteins during this pathological process.
Keywords:
Respiration, artificial; Cytoskeleton; Cell adhesion molecules; Focal adhesions; Mechanotransduction, cellular
RESUMO
A ventilação mecânica é uma terapia importante, mas pode resultar em complicações. Uma das mais relevantes é a lesão pulmonar induzida por ventilador. Devido à hiperdistensão alveolar, o pulmão inicia um processo inflamatório, com infiltrado neutrofílico, formação de membrana hialina, fibrogênese e prejuízo de troca gasosa. Nesse processo, a mecanotransdução da hiperdistensão celular é mediada através do citoesqueleto da célula e de suas interações com a matriz extracelular e com as células vizinhas, de modo que o estímulo mecânico da ventilação se traduz em sinalização bioquímica intracelular, desencadeando ativação endotelial, permeabilidade vascular pulmonar, quimiotaxia leucocitária, produção de citocinas e, possivelmente, lesão de órgãos à distância. Estudos clínicos demonstram essa relação entre distensão pulmonar e mortalidade em pacientes com lesão pulmonar induzida por ventilador. Entretanto, apesar de o citoesqueleto ter um papel fundamental na patogênese da lesão pulmonar induzida por ventilador, a literatura carece de estudos utilizando modelos in vivo sobre as alterações do citoesqueleto e de suas proteínas associadas durante esse processo patológico.
Palavras-chave:
Respiração artificial; Citoesqueleto; Moléculas de adesão celular; Aderências focais; Mecanotransdução celular.
IntroductionMechanical ventilation is a therapy that is widely used in intensive care medicine. The purpose of mechanical ventilation is to provide support when spontaneous ventilation is unable to sustain life or when ventilatory control is imperative in order to prevent the imminent collapse of certain organic functions.(1) Although mechanical ventilation is often indispensable and life-saving, its use has been related to various possible complications, such as increased risk of pneumonia, hemodynamic changes, neuromuscular disorders and barotrauma.(2-5)
Mechanical ventilation can cause direct injury to the lungs, a type of injury known as ventilator-induced lung injury (VILI).(5,6) Initially seen only as a synonym for barotrauma, VILI is currently known to present as nonspecific physiological and morphological changes in the lung parenchyma. Changes in the balance of fluids in the lungs, diffuse alveolar injury, increased endothelial permeability and increased epithelial permeability have been described in animals submitted to mechanical ventilation, leading to a pattern that is often similar to that found in patients with ARDS.(7) Therefore, VILI cannot be distinguished from most of the pulmonary diseases that lead to the use of mechanical ventilation, and VILI can worsen the initial profile.
VILI: from discovery to mechanical determinants
Webb and Tierney were the first authors to describe the pulmonary changes induced by mechanical ventilation with high tidal volume (VT).(8) They studied rats that were ventilated with a positive end-expiratory pressure of 0 cmH2O and a peak inspiratory pressure of 14, 30 or 45 cmH2O. Nonventilated control animals and those submitted to a peak inspiratory pressure of 14 cmH2O presented no pathological changes in the lung. Animals submitted to a peak inspiratory pressure of 30 cmH2O showed perivascular edema without alveolar edema, whereas those submitted to a peak inspiratory pressure of 45 cmH2O showed perivascular edema, alveolar edema, severe hypoxemia and reduced dynamic compliance and died one hour after the beginning of the experiment.(8)
Subsequent studies focused on the mechanical factors that triggered VILI, such as peak inspiratory pressure and VT.(9-12) Dreyfuss et al. confirmed the findings of Webb and Tierney and demonstrated that VT was the major determinant of alveolar injury and alveolocapillary membrane injury, which gave rise to the term "volutrauma".(13,14) In an important study, they compared rats ventilated with high VT and negative pressure (through a negative pressure ventilator, known as iron lung) with rats ventilated with high pressure and low VT (with thoracoabdominal movement restricted by a band). Edema of extravascular lung regions and increased microvascular permeability occurred only in the animals ventilated with high VT, which led to the conclusion that the increase in VT was responsible for the ventilation-induced edema.(13) It should be highlighted that the model used did not allow the authors to distinguish between the deleterious effects of VT and those related to increased transpulmonary pressure.
Based on these data, clinical studies involving lung-protective ventilation strategies were developed with the purpose of minimizing VILI by reducing the VT. The impact of these studies is evident: a reduction in morbidity and a 22% reduction in mortality in patients with ARDS.(15,16)
From volutrauma to biotrauma
Depending on the ventilation strategy adopted, mechanical stress can alter the cellular processes in the lung.(17) Tremblay et al. were some of the first researchers to demonstrate, in an isolated rat lung model, that injurious strategies with high VT induced the production of cytokines (TNF-α) and the expression of c-fos mRNA.(18) Other studies, investigating cultures of cells submitted to cyclic stretching, also demonstrated the production of cytokines in epithelial cells(19) and endothelial cells(20) when these were submitted to a distension of over 80% of the TLC. In addition to being temporally progressive,(21) this cytokine synthesis is potentiated by concomitant factors, such as systemic endotoxins and hyperoxia.(22,23)
Based on the induction of a proinflammatory response in the lung epithelium associated with an alteration in permeability in the local endothelium,(14,24) some authors have suggested that there is decompartmentalization of inflammation, moving to a systemic level. This triggers a distant inflammatory response, favoring multiple organ dysfunction, a phenomenon designated "biotrauma".(25-27)
An important experimental study has demonstrated that injurious mechanical ventilation triggers apoptosis of epithelial cells in the kidney and small intestine, a mechanism that is partially related to the production of Fas ligand.(28) It has also been suggested that part of the organ dysfunction that patients present during the critical period is triggered by ventilation-induced distant apoptosis.
A study evaluating the data regarding IL-6 (a proinflammatory cytokine) corroborated this idea.(16) In that study, the values of IL-6 observed in the group of patients with ARDS who were ventilated with low VT were lower than those observed in the group of patients with ARDS who were ventilated with habitual VT, which suggested decreased inflammatory response due to the ventilation strategy adopted.(16)
However, the biotrauma theory is controversial with regard to the consistency of the production and the relative importance of lung cytokines.(29) This sometimes heated debate remains unsettled.(30,31)
Different tensions act on the lung parenchyma
For several years, it has been known that the distribution of the forces of stress and distension on the lung parenchyma is not uniform.(32) Models of the micromechanics of the parenchyma have revealed that elastic and collagen fibers, helicoidally arranged in the interstitium, are the principal support structures that resist stress.(33-35) In contrast, the alveolar septum is subject to different types of forces and tensions: the endothelial cells are subject to transcapillary pressure and laminar blood flow, whereas the epithelial cells of the airways and alveoli undergo cyclic stretching and compression caused by the respiratory movements and by the surface tension at the air-liquid interface. These forces become even more significant in the presence of parenchymal heterogeneity.(36)
Lung cells need to adapt constantly to the frequent changes in the tension of the tissue in which they are located. The epithelium and endothelium are both subject to various forces during respiration, as a result of the interdependent effects of lung volume, transpulmonary pressure, surface tension and transcapillary tension on the alveolocapillary membrane.(37,38) These forces, at the cellular level, are transmitted and supported by the cytoskeleton of the cell.
The cytoskeleton and its dynamics in cell deformation
The cytoskeleton is composed of an intracellular network of biopolymers that combine to form three types of filaments: microtubules; intermediate filaments; and actin microfilaments. Microtubules are tubulin polymers, which are formed in the perinuclear region and extend to the periphery, conditioning cell shape and guiding intracellular transport. The intermediate filaments are connected to the nuclear envelope, intercellular junctions (such as desmosomes and hemidesmosomes) and other elements of the cytoskeleton, providing the structure with resistance. The intermediate filaments are formed by various proteins, depending on the cell type: in muscle cells, they are formed by desmin; in epithelial cells, they are formed by cytokeratin; and in mesenchymal cells, they are formed by vimentin. Actin microfilaments participate in the generation of contractile forces and are connected to the intracellular proteins of the adherens junctions (in the case of cell-cell contact) or of the focal adhesion plaques (in the case of cell-extracellular matrix contact), stabilizing the cell in relation to the forces that act on the membrane and determining the shape of the plasma membrane.(39)
Being a dynamic and adaptable system, the cytoskeleton has been described as "ant lines": one line can persist for hours if necessary; however, each individual element that constitutes the line is in constant activity. In case there is a new situation, the line can rapidly rearrange itself due to its dynamic nature.(39) In keeping with this metaphor, the cytoskeleton is constantly rearranging itself so that the cell can adapt to the different environmental tensions.
The cytoskeleton is an integrated network, which is why each isolated element cannot be considered for resistance to deformation. According to a biomechanical model designated tensegrity, cells are prestressed structures, composed of a network (the cytoskeleton) of tension-bearing filaments that are coupled to compression-resistant microtubules. This model is stable, resistant and allows cells to respond promptly to external physical stimuli.(40) Since cells are dynamic structures, they actively remodel in order to adapt to the different tensions that act on the lung during respiration. In order to do so, the cytoskeleton, which is the cell structure that is responsible for enduring such deformations and the resulting tensions, degrades and reorganizes itself, providing the system with plasticity. The cytoskeleton has been described as having an amorphous behavior that is dynamic, is similar to that of glass.(41) It is far from being a rigid and static structure, as it was once described. The cytoskeleton is able to restructure itself rapidly, adapting to different tensions and situations; it also changes cell viscosity so that the cell can adapt better to the environment.(42,43)
The cells that constitute the lung epithelium are connected to one another through specialized junctions in the plasma membrane, which are responsible for enduring most of the distension stress. In order to do so, the cytoskeleton filaments insert themselves in these regions, which are rich in cadherins (membrane proteins that are directly responsible for cell-to-cell adhesion). These specialized cell junctions are the adherens junctions and desmosomes, which are in charge of mechanically coupling with the cytoskeleton of neighboring cells.(44)
An example of cytoskeleton reorganization is shown in Figures 1 and 2, in which the structures of adhesion between the alveolar epithelial cells of rats ventilated with low VT (Figure 1) and those of rats ventilated with high VT (Figure 2) are shown by means of electron microscopy.(45)
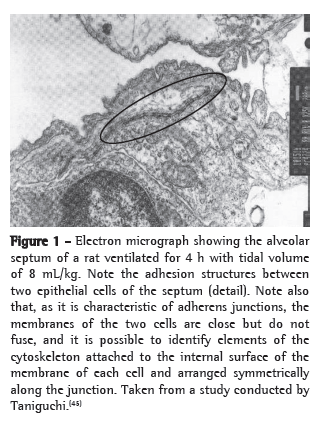
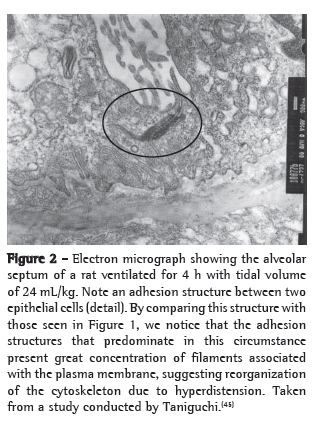
Cell-matrix interaction and mechanotransduction
In addition to connecting to one another, alveolar epithelial cells are connected to the specialized extracellular matrix, known as basal lamina, which supports them.
Integrins are the cell membrane proteins that are responsible for the interaction between the extracellular matrix and the cytoskeleton. The forces that are present in the lung interstitium are transmitted to the cells through the tension on integrins, which retransmit these tensions to the cytoskeleton. An ultrastructural analysis showed the existence of "focal adhesion plaques", which are specialized regions of the cell membrane that are rich in integrins and elements of the cytoskeleton, where the bidirectional interaction between the cell and the matrix occurs.(44) In these adhesion plaques, the bundles of actin microfilaments of the cytoskeleton are anchored to the cytoplasmic face of integrins through a multimolecular complex of junctional proteins. Some of the constituents participate in the structural binding of integrins to the actin cytoskeleton, whereas others are signaling molecules.(46,47) Therefore, these dynamic structures are responsible for monitoring the external environment tension on the cell, playing an important role in cell behavior.(46,47) However, the mechanisms through which the mechanical signs (transmitted to integrins and the cytoskeleton) are transduced into a biochemical sign have yet to be fully understood. Integrins have been reported to generate second intracellular messengers at the anchoring junctions by combining with other proteins, such as paxillin, talin and vinculin, and certain kinases, such as focal adhesion kinase (FAK), PKA, PKC, mitogen-activated protein kinase (MAPK) and c-Src, as well as other enzymatic complexes such as Rho-GTPase and phospholipase C.(48-50)
One of the most important signaling molecules is FAK.(51) A 125-kDa tyrosine kinase, FAK was initially identified in the focal adhesion plaques. It was subsequently demonstrated that, after the interaction between integrin and the extracellular matrix, FAK undergoes a coordinated sequence of events: it is autophosphorylated on tyrosine residues (especially Y397); it combines with other tyrosine kinases of the Src family (such as pp60src); it phosphorylates other proteins associated with the focal adhesion plaques, such as p130Cas and paxillin(52); and it binds to other SH2 proteins, such as Grb2 (in order to activate other signaling pathways, such as Ras and MAPK).(47) Therefore, the location of FAK in the focal adhesion plaque and the phosphorylation of this kinase are important initial events of the mechanotransduction mechanism and play an important role in the formation of new focal junctions.
Some studies have attempted to evaluate the changes in FAK in the processes related to pulmonary hyperdistension. One group of authors studied endothelial cells obtained from an isolated lung model. In that study, the use of high VT led to an increase in FAK phosphorylation and to proinflammatory response in the endothelium, which facilitated leukocyte adhesion by increasing P-selectin.(20) However, another group of authors(53) reported different findings when studying type II pneumocytes isolated from lungs submitted to high VT and hyperoxia. There was a reduction in FAK phosphorylation in these cells, in comparison with those isolated from control rats or with those submitted to high VT without hyperoxia. As a consequence of this reduction in FAK phosphorylation, the ability of cells to adhere to the substrate decreased, demonstrating that this kinase plays an important role in the phenomena of cell-extracellular matrix interaction.(53) Therefore, the different cell types of the lung can respond differently to the same stimulus.
Another important, often investigated, protein is paxillin, which is present in the focal adhesion plaques. This 68-kDa protein does not present catalytic sites in its structure. However, it is rich in binding regions (LD and LIM sites). This suggests a structural and anchoring function for the binding to other proteins. In fact, paxillin is located in adhesion structures that are already established, binding to other structural proteins (such as vinculin and actopaxin), serving as an anchoring structure for enzymes (such as FAK), or a combination of the two.(54) Phosphorylated paxillin has been related to the stabilization and recycling of focal adhesion plaques during the migration and adhesion of cells to the matrix.(55,56) In fact, greater or lower activation of paxillin (via phosphorylation) is believed to be related to a greater or lower migratory potential of the cell, including in the processes of tissue repair.(54)
The findings of studies investigating the behavior of paxillin in VILI are similar to those reported in studies investigating FAK (which is not surprising, since the activation of FAK promotes the phosphorylation of paxillin). Two studies, one investigating the endothelium(20) and the other investigating type II pneumocytes,(53) reported different findings for the phosphorylation of paxillin, the cellular overstretching being similar to that reported in studies investigating FAK. Therefore, the alteration in the phosphorylation of this protein varies according to the cell lineage in question.(54)
In addition to the process of mechanotransduction, new studies have suggested that the cytoskeleton can transmit tensions directly to internal organelles, such as the mitochondria, generating intracellular signs.
Therefore, the hyperdistension on the extracellular matrix associated with the lung endothelium is transmitted to the actin filaments of the cytoskeleton that are connected in series to the integrins and, consequently, to the mitochondria connected to the actin. This triggers a stimulus for the production of free radicals that, among other intracellular effects, promotes P-selectin exocytosis and increases the production of vascular cell adhesion molecule via nuclear factor kappa B (NF-κB), as well as proinflammatory events, which facilitate leukocyte adhesion.(57,58)
In summary, the hyperdistension of the lung parenchyma in VILI seems to be transmitted from the extracellular environment to the interior of the cells through the extracellular matrix-cytoskeleton connection, in which integrin plays the role of mediator. In this mechanism, certain intracellular proteins associated with adhesion structures are important, especially FAK and paxillin. Figure 3 shows a schematic illustration summarizing some of the previously described molecular interactions for signal transduction in the cell adhesion structures.

Mechanisms of independent mechanotransduction of integrins
A mechanism has been proposed by which alveolar hyperdistension can generate intracellular signs that are independent from the cytoskeleton through the formation of transitory fractures in the plasma membrane of cells submitted to mechanical stress. One group of authors demonstrated, in an experimental model, that lungs submitted to high VT showed reversible changes in the plasma membrane.(59) However, these transitory fractures in the membrane can trigger intracellular signaling via c-fos and NF-κB and mediate part of the mechanotransduction independently from the cytoskeleton(60) (Figure 4).
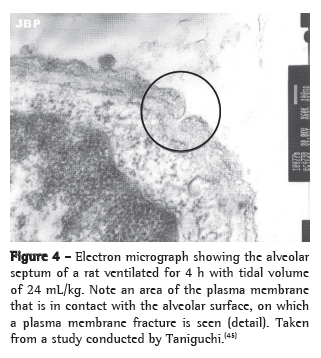
Future perspectives: from in vitro to in vivo models of VILI
As described previously, the cytoskeleton and its associated structures play a crucial role in the pathophysiology of VILI. Studies in this area are progressing toward a better understanding of how mechanotransduction occurs and how the cytoskeleton responds to excessive pulmonary distension. However, these studies have basically focused on cell culture models.(50) Although this reductionist model allows the isolation of each of the components of this complex response, the multiple interactions between the various cell types that constitute the lung are lost. In addition, the culture models submitted to cyclic distension are essentially bidimensional, whereas the alveoli insufflate in the three dimensions. Finally, it cannot be inferred that the impact of the proinflammatory response observed in in vitro models is the same for the body as a whole, especially with regard to the interaction with systemic anti-inflammatory responses, such as IL-10, soluble TNF-α receptor and soluble IL-1 receptor.
There are few in vivo studies of VILI describing the changes in the cytoskeleton or in cytoskeleton-associated proteins, such as FAK and paxillin. Further studies are required in this area, involving models that are more integrated and that can mimic more closely the changes that occur in the pulmonary cells of patients on mechanical ventilation, since these cells are constantly submitted to proinflammatory and pro-apoptotic stimuli, as well as to mechanical stress, which influence their structure and function.
References 1. Hubmayr RD, Irwin RS. Mechanical Ventilation: Initiation. In: Irwin RS, Cerra FB, Rippe JM, editors. Irwin and Rippe's Intensive Care Medicine. Philadelphia: Lippincott-Raven;, 1999. p. 727-41.
2. Kollef MH. Prevention of hospital-associated pneumonia and ventilator-associated pneumonia. Crit Care Med. 2004;32(6):1396-405.
3. Pinsky MR. Recent advances in the clinical application of heart-lung interactions. Curr Opin Crit Care. 2002;8(1):26-31.
4. Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, et al. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med. 2002;166(10):1369-74.
5. Pinhu L, Whitehead T, Evans T, Griffiths M. Ventilator-associated lung injury. Lancet. 2003;361(9354):332-40.
6. Ricard JD, Dreyfuss D, Saumon G. Ventilator-induced lung injury. Eur Respir J Suppl. 2003;42:2s-9s.
7. Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med. 1998;157(1):294-323.
8. Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110(5):556-65.
9. Parker JC, Townsley MI, Rippe B, Taylor AE, Thigpen J. Increased microvascular permeability in dog lungs due to high peak airway pressures. J Appl Physiol. 1984;57(6):1809-16.
10. Hernandez LA, Peevy KJ, Moise AA, Parker JC. Chest wall restriction limits high airway pressure-induced lung injury in young rabbits. J Appl Physiol. 1989;66(5):2364-8.
11. Slutsky AS. Lung injury caused by mechanical ventilation. Chest. 1999;116(1 Suppl):9S-15S.
12. Parker JC, Hernandez LA, Peevy KJ. Mechanisms of ventilator-induced lung injury. Crit Care Med. 1993;21(1):131-43.
13. Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis. 1988;137(5):1159-64.
14. Dreyfuss D, Basset G, Soler P, Saumon G. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis. 1985;132(4):880-4.
15. Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338(6):347-54.
16. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342(18):1301-8.
17. Dos Santos CC, Slutsky AS. Invited review: mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol. 2000;89(4):1645-55.
18. Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997;99(5):944-52.
19. Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol. 1999;277(1 Pt 1):L167-73.
20. Bhattacharya S, Sen N, Yiming MT, Patel R, Parthasarathi K, Quadri S, et al. High tidal volume ventilation induces proinflammatory signaling in rat lung endothelium. Am J Respir Cell Mol Biol. 2003;28(2):218-24.
21. Wilson MR, Choudhury S, Goddard ME, O'Dea KP, Nicholson AG, Takata M. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. J Appl Physiol. 2003;95(4):1385-93.
22. Altemeier WA, Matute-Bello G, Frevert CW, Kawata Y, Kajikawa O, Martin TR, et al. Mechanical ventilation with moderate tidal volumes synergistically increases lung cytokine response to systemic endotoxin. Am J Physiol Lung Cell Mol Physiol. 2004;287(3):L533-42.
23. Quinn DA, Moufarrej RK, Volokhov A, Hales CA. Interactions of lung stretch, hyperoxia, and MIP-2 production in ventilator-induced lung injury. J Appl Physiol. 2002;93(2):517-25.
24. Birukov KG, Jacobson JR, Flores AA, Ye SQ, Birukova AA, Verin AD, et al. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2003;285(4):L785-97.
25. Haitsma JJ, Uhlig S, Göggel R, Verbrugge SJ, Lachmann U, Lachmann B. Ventilator-induced lung injury leads to loss of alveolar and systemic compartmentalization of tumor necrosis factor-alpha. Intensive Care Med. 2000;26(10):1515-22.
26. Tremblay LN, Slutsky AS. Ventilator-induced injury: from barotrauma to biotrauma. Proc Assoc Am Physicians. 1998;110(6):482-8.
27. dos Santos CC, Slutsky AS. Mechanotransduction, ventilator-induced lung injury and multiple organ dysfunction syndrome. Intensive Care Med. 2000;26(5):638-42.
28. Imai Y, Parodo J, Kajikawa O, de Perrot M, Fischer S, Edwards V, et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA. 2003;289(16):2104-12.
29. Ricard JD, Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med. 2001;163(5):1176-80.
30. Dreyfuss D, Ricard JD, Saumon G. On the physiologic and clinical relevance of lung-borne cytokines during ventilator-induced lung injury. Am J Respir Crit Care Med. 2003;167(11):1467-71.
31. Uhlig S, Ranieri M, Slutsky AS. Biotrauma hypothesis of ventilator-induced lung injury. Am J Respir Crit Care Med. 2004;169(2):314-5; author reply 315.
32. Agostoni E. Mechanics of the pleural space. In: Fishman AP, Macklem PT, Mead J, Geiger SR. Handbook of Physiology: Section 3: The Respiratory System Volume III, Parts 1 & 2: Mechanics of Breathing. Baltimore: Williams and Wilkins Co.; 1986. pp. 531-59.
33. Mercer RR, Crapo JD. Spatial distribution of collagen and elastin fibers in the lungs. J Appl Physiol. 1990;69(2):756-65.
34. Oldmixon EH, Hoppin FG Jr. Distribution of elastin and collagen in canine lung alveolar parenchyma. J Appl Physiol. 1989;67(5):1941-9.
35. Mercer RR, Laco JM, Crapo JD. Three-dimensional reconstruction of alveoli in the rat lung for pressure-volume relationships. J Appl Physiol. 1987;62(4):1480-7.
36. Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol. 1970;28(5):596-608.
37. West JB, Tsukimoto K, Mathieu-Costello O, Prediletto R. Stress failure in pulmonary capillaries. J Appl Physiol. 1991;70(4):1731-42.
38. Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med. 2005;171(12):1328-42.
39. Theriot J, Amos L, Cooper J, Fuchs E, Gertler F, Goldstein L, et al. The Cytoskeleton. In: Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, editors. Molecular Biology of the Cell. Nova York: Garland Science;, 2002. p. 907-82.
40. Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol. 1997;59:575-99.
41. Trepat X, Deng L, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, et al. Universal physical responses to stretch in the living cell. Nature. 2007;447(7144):592-5.
42. Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260(5111):1124-7.
43. Gunst SJ, Tang DD, Opazo Saez A. Cytoskeletal remodeling of the airway smooth muscle cell: a mechanism for adaptation to mechanical forces in the lung. Respir Physiol Neurobiol. 2003;137(2-3):151-68.
44. Kypta R, Bernfield M, Burridge K, Geiger B, Goodenough D, Gumbiner B, et al. Cell junctions, cell adhesion, and the extracellular matrix. In: Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, editors. Molecular Biology of the Cell. Nova York: Garland Science;, 2002. p. 1065-125.
45. Taniguchi LU. Avaliação imunohistoquímica das alterações do citoesqueleto na parede alveolar em modelo experimental de lesão pulmonar induzida pela ventilação mecânica em ratos [thesis]. São Paulo: Universidade de São Paulo; 2009.
46. Petit V, Thiery JP. Focal adhesions: structure and dynamics. Biol Cell. 2000;92(7):477-94.
47. Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692(2-3):103-19.
48. Pugin J. Molecular mechanisms of lung cell activation induced by cyclic stretch. Crit Care Med. 2003;31(4 Suppl):S200-6.
49. Han B, Lodyga M, Liu M. Ventilator-induced lung injury: role of protein-protein interaction in mechanosensation. Proc Am Thorac Soc. 2005;2(3):181-7.
50. Liu M, Tanswell AK, Post M. Mechanical force-induced signal transduction in lung cells. Am J Physiol. 1999;277(4 Pt 1):L667-83.
51. Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116(Pt 8):1409-16.
52. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680-8.
53. Desai LP, Sinclair SE, Chapman KE, Hassid A, Waters CM. High tidal volume mechanical ventilation with hyperoxia alters alveolar type II cell adhesion. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L769-78.
54. Brown MC, Turner CE. Paxillin: adapting to change. Physiol Rev. 2004;84(4):1315-39.
55. Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci. 2007;120(Pt 1):137-48.
56. Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001;20(44):6459-72.
57. Ichimura H, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Invest. 2003;111(5):691-9.
58. Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287(3):L486-96.
59. Gajic O, Lee J, Doerr CH, Berrios JC, Myers JL, Hubmayr RD. Ventilator-induced cell wounding and repair in the intact lung. Am J Respir Crit Care Med. 2003;167(8):1057-63.
60. Grembowicz KP, Sprague D, McNeil PL. Temporary disruption of the plasma membrane is required for c-fos expression in response to mechanical stress. Mol Biol Cell. 1999;10(4):1247-57.
* Study carried out at the Faculdade de Medicina da Universidade de São Paulo - FMUSP, University of São Paulo School of Medicine - Hospital das Clínicas, São Paulo, Brazil.
Correspondence to: Leandro Utino Taniguchi. Disciplina de Emergências Clínicas, Hospital das Clínicas da Faculdade de Medicina da USP, Avenida Enéas de Carvalho Aguiar, 155, 8º andar, Bloco 03, Clínica Médica Pós-Graduação Ciências Médicas, CEP 05403-900, São Paulo, SP, Brasil.
Tel 55 11 3069-6336. E-mail: leandrout@hotmail.com
Financial support: This study received financial support from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, National Council for Scientific and Technological Development) and the Laboratories for Medical Research of the University of São Paulo School of Medicine Hospital das Clínicas.
Submitted 5 November 2009. Accepted, after review, 26 January 2010.
About the authorsLeandro Utino Taniguchi
Attending Physician. ICU of the Clinical Medicine Emergency Room, Faculdade de Medicina da Universidade de São Paulo - FMUSP, University of São Paulo School of Medicine - Hospital das Clínicas, São Paulo, Brazil.
Elia Garcia Caldini
Professor. Department of Pathology, Faculdade de Medicina da Universidade de São Paulo - FMUSP, University of São Paulo School of Medicine - São Paulo, Brazil.
Irineu Tadeu Velasco
Full Professor. Department of Clinical Emergencies, Faculdade de Medicina da Universidade de São Paulo - FMUSP, University of São Paulo School of Medicine - São Paulo, Brazil.
Elnara Márcia Negri
Tenured Professor. Faculdade de Medicina da Universidade de São Paulo - FMUSP, University of São Paulo School of Medicine - São Paulo, Brazil.













 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket