ABSTRACT
Objective: To study the expression of COX-1 and COX-2 in the remodeled lung in systemic sclerosis (SSc) and idiopathic pulmonary fibrosis (IPF) patients, correlating that expression with patient survival. Methods: We examined open lung biopsy specimens from 24 SSc patients and 30 IPF patients, using normal lung tissue as a control. The histological patterns included fibrotic nonspecific interstitial pneumonia (NSIP) in SSc patients and usual interstitial pneumonia (UIP) in IPF patients. We used immunohistochemistry and histomorphometry to evaluate the expression of COX-1 and COX-2 in alveolar septa, vessels, and bronchioles. We then correlated that expression with pulmonary function test results and evaluated its impact on patient survival. Results: The expression of COX-1 and COX-2 in alveolar septa was significantly higher in IPF-UIP and SSc-NSIP lung tissue than in the control tissue. No difference was found between IPF-UIP and SSc-NSIP tissue regarding COX-1 and COX-2 expression. Multivariate analysis based on the Cox regression model showed that the factors associated with a low risk of death were younger age, high DLCO/alveolar volume, IPF, and high COX-1 expression in alveolar septa, whereas those associated with a high risk of death were advanced age, low DLCO/alveolar volume, SSc (with NSIP), and low COX-1 expression in alveolar septa. Conclusions: Our findings suggest that strategies aimed at preventing low COX-1 synthesis will have a greater impact on SSc, whereas those aimed at preventing high COX-2 synthesis will have a greater impact on IPF. However, prospective randomized clinical trials are needed in order to confirm that.
Keywords:
Scleroderma, systemic; Idiopathic pulmonary fibrosis; Inflammation; Survival rate.
RESUMO
Objetivo: Estudar a expressão de COX-1 e COX-2 em áreas pulmonares remodeladas em pacientes com esclerose sistêmica (ES) ou fibrose pulmonar idiopática (FPI) e correlacioná-la com a sobrevida desses pacientes. Métodos: Examinamos espécimes de biópsia pulmonar a céu aberto de 24 pacientes com ES e de 30 pacientes com FPI, utilizando-se tecido pulmonar normal como controle. Os padrões histológicos incluíram pneumonia intersticial não específica (PINE) fibrótica em pacientes com ES e pneumonia intersticial usual (PIU) nos pacientes com FPI. Imuno-histoquímica e histomorfometria foram usadas para avaliar a expressão celular de COX-1 e COX-2 em septos alveolares, vasos e bronquíolos, sua correlação com provas de função pulmonar e seu impacto na sobrevida. Resultados: A expressão de COX-1 e COX-2 em septos alveolares foi significativamente maior em FPI-PIU e ES-PINE do que no tecido controle. Não houve diferença entre FPI-PIU e ES-PINE quanto à expressão de COX-1 e COX-2. A análise multivariada baseada no modelo de regressão de Cox mostrou que os fatores associados a baixo risco de morte foram ter idade menor, valores elevados de DLCO/volume alveolar, FPI, e alta expressão de COX-1 em septos alveolares, ao passo que os fatores associados a alto risco de morte foram ter idade maior, valores baixos de DLCO/volume alveolar, ES (com PINE) e baixa expressão de COX-1 em septos alveolares. Conclusões: Nossos resultados sugerem que estratégias de prevenção de baixa síntese de COX-1 terão maior impacto sobre a ES, ao passo que as de prevenção de alta síntese de COX-2 terão maior impacto sobre a FPI. Porém, são necessários ensaios clínicos randomizados prospectivos para confirmar essa hipótese.
Palavras-chave:
Escleroderma sistêmico; Fibrose pulmonar idiopática; Inflamação; Taxa de sobrevida.
IntroduçãoO remodelamento pulmonar é uma sequela terminal comum da fibrose pulmonar idiopática (FPI) e da esclerose sistêmica (ES); resulta em desorganização da arquitetura pulmonar e, consequentemente, insuficiência respiratória progressiva.(1-4) Histologicamente, o processo de remodelamento é caracterizado por inflamação intersticial crônica difusa e aumento da proliferação de fibroblastos, da síntese de matriz extracelular e da deposição de colágeno.(2,5,6) Portanto, a modulação da inflamação, da proliferação de fibroblastos e da síntese de colágeno por mediadores efetores na FPI e na ES é muito importante. Não obstante a caracterização de vários participantes cruciais, os mediadores e mecanismos envolvidos na patogênese da FPI e da ES ainda não foram totalmente definidos, o que pode explicar o número limitado de opções terapêuticas, com pouco impacto na sobrevida em longo prazo.(7,8)
Sabe-se que a COX é a enzima essencial para a conversão de ácido araquidônico em prostaglandina E2 (PGE2), que é o precursor de uma família de mediadores lipídicos bioativos diversos, como as prostaglandinas, o tromboxano e a prostaciclina. A COX existe em duas isoformas: COX-1 e COX-2. A COX-1 é expressa constitutivamente na maioria dos tecidos e atua como uma enzima "que mantém a casa em ordem", isto é, que regula a homeostase vascular, protege a mucosa gástrica e mantém a integridade renal,(9,10) ao passo que a COX-2 tem níveis mais baixos de expressão na maioria dos tecidos, embora possa ser induzida em resposta a fatores de crescimento, citocinas e outras moléculas pró-inflamatórias.(11-13)
No tocante ao papel pró-inflamatório e anti-inflamatório da COX-1 e da COX-2, a imuno-histoquímica pode ser útil para detectar essas enzimas no pulmão remodelado de pacientes com ES e FPI. Dados relativos à avaliação de COX-1 e COX-2 no pulmão remodelado foram anteriormente relatados no soro(14,15) e lavado broncoalveolar(16) de pacientes com ES, bem como em culturas de fibroblastos(4) e biópsias(17,18) de pacientes com FPI. No entanto, o papel da COX-1 e da COX-2 nos mecanismos envolvidos no remodelamento pulmonar em pacientes com FPI e ES ainda não está claro, e há incerteza sobre a melhor maneira de detectar a COX-2. O objetivo do presente estudo foi estudar a expressão de COX-1 e COX-2 (avaliada separadamente em septos alveolares, bronquíolos e vasos) em espécimes obtidos por meio de biópsia pulmonar e correlacioná-la com a sobrevida dos pacientes.
MétodosEntre janeiro de 2002 e julho de 2008, 24 pacientes consecutivos com ES e doença pulmonar intersticial e 30 pacientes com suspeita de FPI baseada em achados de TCAR foram submetidos a biópsia pulmonar a céu aberto no Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, na cidade de São Paulo (SP). Todos os pacientes preencheram os critérios para o diagnóstico de ES(19) e FPI.(1) A biópsia pulmonar a céu aberto foi realizada por meio de toracotomia formal, evitando-se áreas de faveolamento. Todos os pacientes assinaram um termo de consentimento livre e esclarecido, e o estudo foi aprovado pelo comitê de ética em pesquisa da instituição (Protocolo nº 0960/08).
Foram analisados os prontuários clínicos de todos os pacientes. A duração da doença foi determinada com base no início do primeiro sintoma. Os testes de função pulmonar e a TCAR foram realizados em um prazo de até 3 meses antes da biópsia. Os testes de função pulmonar incluíram CV, VEF1, CVF, VEF1/CVF, CPT, VR e DLCO. A avaliação fisiológica foi realizada antes da biópsia pulmonar a céu aberto e antes do início do tratamento. Todos os testes de função pulmonar, incluindo espirometria, determinação dos volumes pulmonares e medição da DLCO, foram realizados no mesmo dia. Todos os testes espirométricos foram realizados com um pneumotacógrafo calibrado (Medical Graphics Co., St. Paul, MN, EUA); todos os valores foram expressos em porcentagem de seus respectivos valores previstos, os valores de referência tendo sido estabelecidos por Pereira et al.(20) Os volumes pulmonares foram medidos com um pletismógrafo de corpo inteiro (Medical Graphics Co.); todos os valores foram expressos em porcentagem dos valores previstos. (21) A capacidade de difusão foi expressa em porcentagem dos valores previstos. (22) Todos os pacientes foram acompanhados regularmente após o tratamento até o óbito; exames de sangue e testes de função pulmonar foram realizados regularmente. O desfecho primário foi avaliar o impacto de alterações em COX-1 e COX-2 na sobrevida e analisar as diferenças entre ES e FPI. A Tabela 1 mostra os dados demográficos. Como controle, obtivemos tecido pulmonar normal de 10 indivíduos (6 homens e 4 mulheres) cuja mediana de idade foi de 46,6 5,8 anos e que morreram subitamente de causas não pulmonares.
Quanto aos achados da biópsia pulmonar a céu aberto, a pneumonia intersticial usual (PIU), o padrão histológico da FPI, foi caracterizada por distribuição subpleural e parasseptal segmentada de lesões parenquimatosas. Com um aumento pequeno, observou-se heterogeneidade temporal, com áreas de parênquima pulmonar normal se alternando com colapso alveolar, infiltrado mononuclear intersticial, tecido fibromixoide septal (focos fibroblásticos) e faveolamento.(2) Todos os pacientes com ES apresentavam padrões histológicos consistentes com pneumonia intersticial não específica (PINE) fibrótica, definida por espessamento septal temporalmente homogêneo e fibrose intersticial.(19)
Para a análise imuno-histoquímica, foi usada uma técnica-padrão de peroxidase - com a hematoxilina de Harris como contracorante - a fim de identificar a expressão de COX-1 e COX-2 em septos alveolares, paredes bronquiolares e paredes vasculares em tecido pulmonar normal (o tecido controle), em tecido pulmonar com padrão histológico de PIU (o tecido PIU) e em tecido pulmonar com padrão histológico de PINE (o tecido PINE). Todos os anticorpos usados foram anticorpos policlonais de cabra biotinilados. Anticorpos anti-COX-1 e anti-COX-2 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, EUA) foram incubados com cortes histológicos em diluições de 1:50 e 1:100, respectivamente. O kit de amplificação Novolink Max Polymer (Leica Biosystems Newcastle Ltd, Newcastle upon Tyne, Reino Unido) foi usado para a amplificação do sinal, e 3,3'-diaminobenzidina tetra-hidrocloreto (0,25 mg dissolvidos em 1 mL de peróxido de hidrogênio a 0,02%) foi usado como substrato precipitante para a detecção do sinal. A especificidade dos anticorpos primários foi confirmada por controles de reagentes apropriados - o anticorpo primário sendo omitido ou soro não imune sendo usado em vez do anticorpo primário no protocolo de coloração - os quais não revelaram nenhuma coloração.
Quanto à histomorfometria, avaliamos a expressão de COX-1 e COX-2 por meio de uma técnica de contagem de pontos em 50 e 30 campos em septos alveolares, paredes bronquiolares, e paredes vasculares no tecido controle, no tecido PIU e no tecido PINE. A técnica foi executada com um retículo de 100 pontos (área: 187.500 m(21); aumento: 400×) ligado à ocular do microscópio.(23) Com um aumento de 400×, as áreas septais, bronquiolares e vasculares em cada campo foram calculadas com base no número de pontos sobrepostos ao tecido conjuntivo, na forma de porcentagem da área total do retículo. Posteriormente, contou-se o número de células imunomarcadas nas áreas septais, bronquiolares e vasculares. A fração de área de células imunomarcadas representa a relação percentual entre a área de células marcadas e a área total coberta pelo retículo na ocular.
Para avaliar a variabilidade interobservador, comparamos os resultados obtidos por dois observadores em 20% das lâminas. O coeficiente de variação para o erro interobservador da contagem de células foi de 5%.
Os dados são apresentados na forma de média dp e IC95%. O teste t de Student para amostras independentes foi usado a fim de testar a relação entre variáveis contínuas, e os resíduos foram examinados para certificar que sua distribuição era aproximadamente normal. A relação entre celularidade (determinada pela imunomarcação) e os resultados dos testes de função pulmonar foi avaliada pelo coeficiente de correlação de Pearson. Para todos os casos, os valores das variáveis medidas foram dispostos em ordem crescente e divididos em dois grupos com base na mediana de cada variável. Para cada variável, os grupos foram denominados baixo grau e alto grau, a saber: COX-1 em septos alveolares (baixo grau: < 2,35%; alto grau: 2,35%); COX-1 em vasos (baixo grau: < 2,91%; alto grau: 2,91%); COX-1 em bronquíolos (baixo grau: < 2,88%; alto grau: 2,88%); COX-1 total (baixo grau: < 2,77%; alto grau: 2,77%); COX-2 em septos alveolares (baixo grau: < 2,04%; alto grau: 2,04%); COX-2 em vasos (baixo grau: < 2,34%; alto grau: 2,34%); COX-2 em bronquíolos (baixo grau: < 2,34%; alto grau: 2,34%) e COX-2 total (baixo grau: < 2,16%; alto grau: 2,16%).
A análise da sobrevida global foi realizada em dois passos. Primeiro, realizamos uma análise univariada relacionando o acompanhamento global com cada uma das variáveis medidas por meio do método de Kaplan-Meier e então analisamos a sobrevida por meio do teste de log-rank. As variáveis que se mostraram significantes na análise univariada foram incluídas na análise multivariada baseada no modelo de regressão de riscos proporcionais de Cox. Qualquer morte causada por FPI ou ES foi considerada um evento positivo. Mortes cujas causas foram outras que não FPI ou ES e pacientes vivos foram incluídos nos modelos como casos censurados.
Todos os procedimentos estatísticos foram realizados com o programa Statistical Package for the Social Sciences, versão 18.0 (SPSS Inc., Chicago, IL, EUA). Para todos os testes, o nível de significância adotado foi de 5%.
ResultadosA Tabela 1 resume as características clínicas dos pacientes com ES (n = 24) e dos pacientes com FPI (n = 30). Seis de 17 pacientes com ES (35,29%) e 13 de 19 pacientes com FPI (68,42%) apresentaram distúrbio ventilatório restritivo. Os resultados dos testes de função respiratória foram os seguintes: CVF < 80% em 18 (75%) dos 24 pacientes com ES e em 19 de 22 pacientes com FPI (86,36%); CPT < 80% em 6 de 17 pacientes com ES (35,9%) e em 13 de 19 pacientes com FPI (68,42%); DLCO < 80% em 12 de 15 pacientes com ES (80%) e em 8 de 9 pacientes com FPI (88,88%); DLCO/volume alveolar < 80% em 11 de 18 pacientes com ES (61,11%) e em 11 de 14 pacientes com FPI (78,57%). Houve correlação negativa significativa entre a expressão de COX-2 em vasos e a CVF (r = −0,28; p = 0,05) e entre a expressão de COX-2 em septos alveolares e a DLCO (r = −0,80; p = 0,009).

A Figura 1 mostra septos alveolares, vasos e bronquíolos no tecido controle, no tecido PINE e no tecido PIU com imunomarcação positiva para COX-1 (em A, C, E, G, I, K, M, O e Q) e COX-2 (em B, D, F, H, J, L, N, P e R). Os tecidos PINE e PIU diferiram do tecido controle quanto à intensidade da imunomarcação de células epiteliais, células endoteliais, miofibroblastos e células musculares lisas nos septos alveolares, vasos, e bronquíolos.
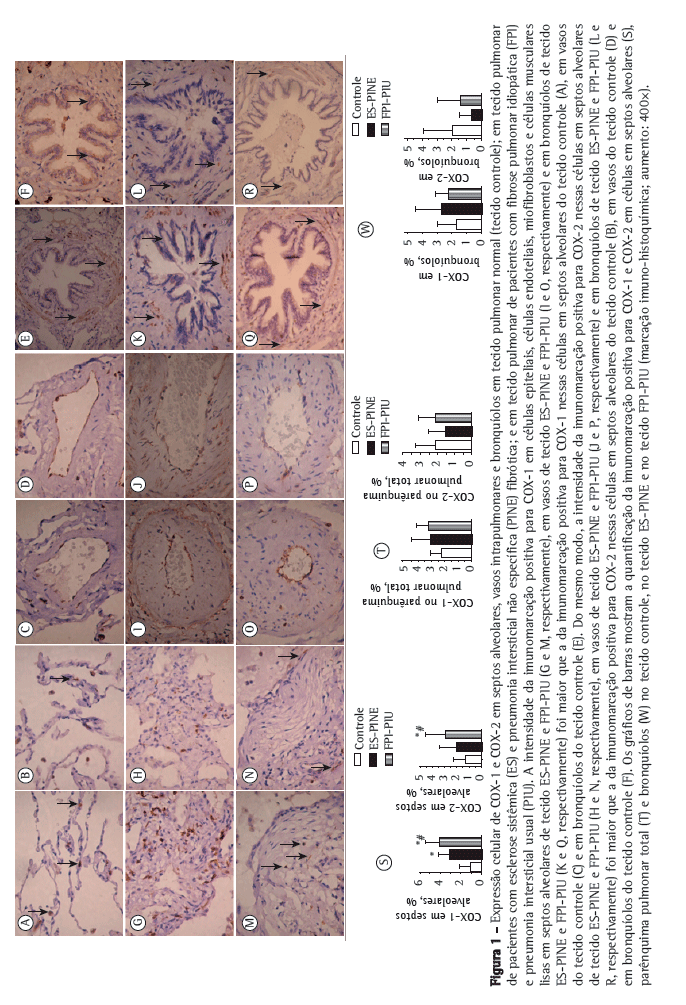
A Tabela 2 resume os resultados morfométricos. A proporção de células em septos alveolares com imunomarcação positiva para COX-1 e COX-2 foi significativamente maior nos tecidos PIU e PINE do que no tecido controle. Em outras palavras, elevadas proporções de células em septos alveolares marcadas positivamente para COX-1 e COX-2 associaram-se aos padrões histológicos de PIU e PINE. Como se pode observar nos gráficos de barras na Figura 1 (S e T), a relação de COX-1 e COX-2 com a FPI (o padrão histológico de PIU) foi mais forte do que a de COX-1 e COX-2 com a ES (o padrão histológico de PINE). Embora a proporção de células bronquiolares com imunomarcação positiva para COX-2 tenha sido menor nos tecidos PINE e PIU do que no tecido controle (Figura 1W), a diferença não foi estatisticamente significante. Além disso, embora a proporção de células bronquiolares com imunomarcação positiva para COX-1 tenha sido maior nos tecidos PIU e PINE do que no tecido controle (Figura 1W), a diferença não foi significante. Não foram encontradas diferenças entre os tecidos no tocante à imunomarcação de COX-1 e COX-2 em vasos e no parênquima total (Tabela 2).

Uma análise preliminar das curvas de sobrevida de Kaplan-Meier mostrou que a sobrevida foi melhor nos pacientes com ES (o padrão histológico de PINE fibrótica) e expressão de COX-2 > 2,25% (mediana da sobrevida: 70,75 meses) do que naqueles com FPI (o padrão histológico de PIU) e expressão de COX-2 < 2,25% (mediana da sobrevida: 46,32 meses; Figura 2). Portanto, codificamos o padrão histológico de PINE fibrótica como uma única variável dummy com valor = 1 e o padrão histológico de PIU com valor = 2. Os resultados da análise multivariada baseada no modelo de regressão de riscos proporcionais de Cox são apresentados na Tabela 3. Após termos controlado a idade, os resultados dos testes de função pulmonar, o padrão histológico de PIU e o padrão histológico de PINE fibrótica, constatamos que apenas duas variáveis se associaram de maneira significativa ao tempo de sobrevida: o padrão histológico de PINE fibrótica e COX-2 em septos alveolares (p = 0,02). Uma vez que essas duas variáveis foram contabilizadas, nenhuma das demais se relacionou à sobrevida. A análise multivariada revelou baixo risco de morte para pacientes jovens com VEF1/CVF baixa, padrão histológico de PINE fibrótica e COX-2 de alto grau em septos alveolares.

 Discussão
DiscussãoO número limitado de abordagens terapêuticas que tenham qualquer impacto na sobrevida em longo prazo em pacientes com FPI-PIU e naqueles com ES e PINE fibrótica deve-se à falta de definição a respeito dos mediadores e mecanismos envolvidos na patogênese da FPI e da ES. Portanto, a questão de interesse é se mediadores adicionais podem ajudar a compreender melhor a patogênese dessas doenças. O processo de reparo envolve duas fases distintas: uma fase inflamatória regenerativa, em que o microambiente tenta substituir as células lesadas, e uma fase fibrótica, em que o tecido conjuntivo substitui o tecido parenquimatoso normal.(24-26) No processo de reparo, aumenta a produção de PGE2 por fibroblastos,(27,28) o que constitui evidência adicional das propriedades antiproliferativas, anti-inflamatórias e antifibróticas da COX-2/PGE2.(15) Portanto, nosso achado de que a marcação imuno-histoquímica para COX fornece informações importantes sobre os processos de reparo na fibrose pulmonar não é surpreendente, e nossos resultados confirmam que a expressão de COX-2 é maior na FPI e na ES, com desfecho melhor em um grupo de pacientes. Constatamos que a proporção de células em septos alveolares com imunomarcação positiva para COX-1 e COX-2 foi significativamente maior em tecido pulmonar com padrão histológico de PIU e PINE fibrótica do que em tecido pulmonar normal. A maior expressão de COX-1 era esperada, pois a COX-1 é expressa constitutivamente na maioria das células e tecidos, ao passo que a COX-2 é induzida por estímulos inflamatórios ou mitogênicos.(9) Esses resultados contrastam com os de estudos anteriores nos quais se investigou a FPI.(4,17,18) Esses estudos mostraram uma redução da expressão de COX-2 em fibroblastos pulmonares em consequência de uma diminuição da produção de COX-2. No entanto, nesses estudos, a expressão de COX-2 foi medida apenas em fibroblastos, ao passo que em nosso estudo ela foi medida nos septos alveolares, incluindo células epiteliais e fibroblastos em áreas normais, áreas em colapso, e focos de fibroblastos. Outros estudos, inclusive um estudo realizado por Lappi-Blanco et al.,(3) encontraram um aumento da expressão de COX-2 em epitélio metaplásico e fibroblastos provenientes de áreas fibróticas em FPI-PIU. Esses resultados divergentes sugerem que a COX-2 desempenha um papel duplo na FPI-PIU. Primeiro, a redução da expressão da COX-2 em áreas normais, áreas em colapso e focos fibroblásticos sugere que a COX-2 desempenha um papel anti-inflamatório na FPI-PIU em fase inicial. Segundo, a presença de fibrose progressiva mesmo na presença de aumento da expressão de COX-2 sugere que os fibroblastos são incapazes de responder ao estímulo da COX-2 e seu produto principal (PGE2) de modo a inibir a proliferação de fibroblastos, a transformação miofibroblástica e o aumento da produção de colágeno e outras moléculas da matriz extracelular.
No presente estudo, a proporção de células em septos alveolares com imunomarcação positiva para COX-1 e COX-2 foi menor em tecido fibrótico PINE (proveniente de pacientes com ES) do que em tecido PIU (de pacientes com FPI). Esse achado contrasta com os de estudos anteriores em que se mostrou que os níveis de COX-2 são mais elevados em pacientes com ES.(14-16) Além disso, relatou-se que a produção de COX-2 é muito maior na fase de resolução inflamatória do que na fase inicial.(29) Esses achados divergentes sugerem que a COX-2 tem um papel duplo em um processo inflamatório normal, desempenhando um papel pró-inflamatório na fase inicial e um papel anti-inflamatório na fase de resolução.(29) Portanto, diante das evidências mencionadas acima e da inflamação latente em pacientes com ES e envolvimento pulmonar, nossos resultados reforçam a ideia de que a COX-2 não exerce seu efeito anti-inflamatório de maneira apropriada, pois há inflamação mesmo quando a expressão de COX-2 está aumentada em pacientes com ES e PINE fibrótica. No entanto, mais estudos são necessários para esclarecer o verdadeiro motivo pelo qual o mecanismo da COX-2 é deficiente. Supomos que isso se deva à incapacidade da COX-2 de estimular a produção de PGE2 ou de outros mediadores anti-inflamatórios em oposição a seus próprios efeitos pró-inflamatórios ou à incapacidade das células de reagir apropriadamente à COX-2.
Nosso estudo tem impacto clínico e funcional. Procuramos estabelecer uma correlação entre a COX-2 e a sobrevida dos pacientes controlada quanto à idade, aos resultados dos testes de função pulmonar, ao padrão histológico de PIU (em pacientes com FPI) e ao padrão histológico de PINE (em pacientes com ES). Nossa análise multivariada revelou baixo risco de morte para pacientes mais jovens com DLCO/volume alveolar baixa, ES (e o padrão histológico de PINE) COX-2 total de alto grau e COX-1 de alto grau em septos alveolares.
Em suma, a expressão de COX-1 e COX-2 no parênquima pulmonar nos oferece o potencial de controlar processos de reparo envolvidos na progressão da ES-PINE e da FPI-PIU, sugerindo que estratégias destinadas a prevenir a baixa síntese de COX-1 terão maior impacto na ES, ao passo que as destinadas a prevenir a elevada síntese de COX-2 terão maior impacto na FPI. Ensaios randomizados prospectivos são necessários para confirmar isso.
Referências1. Sociedade Brasileira de Pneumologia e Tisiologia. Diretrizes de Doenças Pulmonares Intersticiais da Sociedade Brasileira de Pneumologia e Tisiologia. J Bras Pneumol. 2012;38(Suppl 2):S1-S133.
2. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. American Thoracic Society; European Respiratory Society. Am J Respir Crit Care Med. 2002;165(2):277-304. Erratum in: Am J Respir Crit Care Med. 2002;166(3):426. http://dx.doi.org/10.1164/ajrccm.165.2.ats01 PMid:11790668
3. Lappi-Blanco E, Kaarteenaho-Wiik R, Maasilta PK, Anttila S, Pääkkö P, Wolff HJ. COX-2 is widely expressed in metaplastic epithelium in pulmonary fibrous disorders. Am J Clin Pathol. 2006;126(5):717-24. http://dx.doi.org/10.1309/PFGXCLNG2N17PJX9 PMid:17050069
4. Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest. 1995;95(4):1861-8. http://dx.doi.org/10.1172/JCI117866 PMid:7706493 PMCid:PMC295728
5. Gabbiani G. The myofibroblast: a key cell for wound healing and fibrocontractive diseases. Prog Clin Biol Res. 1981;54:183-94. PMid:7015359
6. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345(7):517-25. http://dx.doi.org/10.1056/NEJMra003200 PMid:11519507
7. Dempsey OJ. Clinical review: idiopathic pulmonary fibrosis--past, present and future. Respir Med. 2006;100(11):1871-85. http://dx.doi.org/10.1016/j.rmed.2006.08.017 PMid:16987645
8. Downey DG, Brockbank S, Martin SL, Ennis M, Elborn JS. The effect of treatment of cystic fibrosis pulmonary exacerbations on airways and systemic inflammation. Pediatr Pulmonol. 2007;42(8):729-35. http://dx.doi.org/10.1002/ppul.20646 PMid:17588254
9. Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12(12):1063-73. PMid:9737710
10. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97-120. http://dx.doi.org/10.1146/annurev.pharmtox.38.1.97 PMid:9597150
11. Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A. 1991;88(7):2692-6. http://dx.doi.org/10.1073/pnas.88.7.2692 PMid:1849272 PMCid:PMC51304
12. O'Banion MK, Sadowski HB, Winn V, Young DA. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. J Biol Chem. 1991;266(34):23261-7. PMid:1744122
13. Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A. 1992;89(16):7384-8. http://dx.doi.org/10.1073/pnas.89.16.7384
14. Bassyouni IH, Talaat RM, Salem TA. Serum concentrations of cyclooxygenase-2 in patients with systemic sclerosis: association with lower frequency of pulmonary fibrosis. J Clin Immunol. 2012;32(1):124-30. http://dx.doi.org/10.1007/s10875-011-9601-z PMid:21979415
15. Xiao R, Kanekura T, Yoshida N, Higashi Y, Yan KL, Fukushige T, et al. 9-Cis-retinoic acid exhibits antifibrotic activity via the induction of cyclooxygenase-2 expression and prostaglandin E2 production in scleroderma fibroblasts. Clin Exp Dermatol. 2008;33(4):484-90. http://dx.doi.org/10.1111/j.1365-2230.2008.02727.x PMid:18462443
16. Kowal-Bielecka O, Kowal K, Distler O, Rojewska J, Bodzenta-Lukaszyk A, Michel BA, et al. Cyclooxygenase- and lipoxygenase-derived eicosanoids in bronchoalveolar lavage fluid from patients with scleroderma lung disease: an imbalance between proinflammatory and antiinflammatory lipid mediators. Arthritis Rheum. 2005;52(12):3783-91. http://dx.doi.org/10.1002/art.21432 PMid:16320329
17. Xaubet A, Roca-Ferrer J, Pujols L, Ramírez J, Mullol J, Marin-Arguedas A, et al. Cyclooxygenase-2 is up-regulated in lung parenchyma of chronic obstructive pulmonary disease and down-regulated in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21(1):35-42. PMid:15127973
18. Petkova DK, Clelland CA, Ronan JE, Lewis S, Knox AJ. Reduced expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis. Histopathology. 2003;43(4):381-6. http://dx.doi.org/10.1046/j.1365-2559.2003.01718.x PMid:14511257
19. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202-5. PMid:3361530
20. Pereira CA, Sato T, Rodrigues SC. New reference values for forced spirometry in white adults in Brazil. J Bras Pneumol. 2007;33(4):397-406. http://dx.doi.org/10.1590/S1806-37132007000400008 PMid:17982531
21. Neder JA, Andreoni S, Castelo-Filho A, Nery LE. Reference values for lung function tests. I. Static volumes. Braz J Med Biol Res. 1999;32(6):703-17. PMid:10412549
22. Andreoni S, Lerario MC, Nery LE. Reference values for lung function tests. II. Maximal respiratory pressures and voluntary ventilation. Braz J Med Biol Res. 1999;32(6):719-27. PMid:10412550
23. Hsia CC, Hyde DM, Ochs M, Weibel ER; ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181(4):394-418. http://dx.doi.org/10.1164/rccm.200809-1522ST PMid:20130146
24. Meneghin A, Hogaboam CM. Infectious disease, the innate immune response, and fibrosis. J Clin Invest. 2007;117(3):530-8. http://dx.doi.org/10.1172/JCI30595 PMid:17332880 PMCid:PMC1804377
25. Oikonomou N, Harokopos V, Zalevsky J, Valavanis C, Kotanidou A, Szymkowski DE, et al. Soluble TNF mediates the transition from pulmonary inflammation to fibrosis. PLoS One. 2006;1:e108. http://dx.doi.org/10.1371/journal.pone.0000108 PMid:17205112 PMCid:PMC1762410
26. Pardo A, Selman M. Molecular mechanisms of pulmonary fibrosis. Front Biosci. 2002;7:d1743-61.
27. Kobayashi T, Narumiya S. Function of prostanoid receptors: studies on knockout mice. Prostaglandins Other Lipid Mediat. 2002;68-69:557-73. http://dx.doi.org/10.1016/S0090-6980(02)00055-2
28. McCann MR, Monemdjou R, Ghassemi-Kakroodi P, Fahmi H, Perez G, Liu S, et al. mPGES-1 null mice are resistant to bleomycin-induced skin fibrosis. Arthritis Res Ther. 2011;13(1):R6. http://dx.doi.org/10.1186/ar3226 PMid:21266028 PMCid:PMC3546456
29. Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5(6):698-701. http://dx.doi.org/10.1038/9550 PMid:10371510
Sobre os autoresEdwin Roger Parra
Pós-doutorando. Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.
Flavia Lin
Estudante de Medicina. Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.
Vanessa Martins
Mestranda. Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.
Maristela Peres Rangel
Doutoranda. Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.
Vera Luiza Capelozzi
Professora Associada. Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo (SP) Brasil.






 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket




