ABSTRACT
Objective: To determine the frequency of six mutations (F508del, G542X, G551D, R553X, R1162X, and N1303K) in patients with cystic fibrosis (CF) diagnosed, at a referral center, on the basis of abnormal results in two determinations of sweat sodium and chloride concentrations. Methods: This was a cross-sectional study involving 70 patients with CF. The mean age of the patients was 12.38 ± 9.00 years, 51.43% were female, and 94.29% were White. Mutation screening was performed with polymerase chain reaction (for F508del), followed by enzymatic digestion (for other mutations). Clinical analysis was performed on the basis of gender, age, ethnicity, pulmonary/gastrointestinal symptoms, and Shwachman-Kulczycki (SK) score. Results: All of the patients showed pulmonary symptoms, and 8 had no gastrointestinal symptoms. On the basis of the SK scores, CF was determined to be mild, moderate, and severe in 22 (42.3%), 17 (32.7%), and 13 (25.0%) of the patients, respectively. There was no association between F508del mutation and disease severity by SK score. Of the 140 alleles analyzed, F508del mutation was identified in 70 (50%). Other mutations (G542X, G551D, R553X, R1162X, and N1303K) were identified in 12 (7.93%) of the alleles studied. In F508del homozygous patients with severe disease, the OR was 0.124 (95% CI: 0.005-0.826). Conclusions: In 50% of the alleles studied, the molecular diagnosis of CF was confirmed by identifying a single mutation (F508del). If we consider the analysis of the six most common mutations in the Brazilian population (including F508del), the molecular diagnosis was confirmed in 58.57% of the alleles studied.
RESUMO
Objetivo: Determinar a frequência de seis mutações (F508del, G542X, G551D, R553X, R1162X e N1303K) em pacientes com fibrose cística (FC) de um centro de referência, diagnosticados pela presença de duas dosagens de sódio e cloro no suor alteradas. Métodos: Estudo de corte transversal com 70 pacientes com idade média de 12,38 ± 9,00 anos, sendo que 51,43% eram do sexo feminino, e 94,29% eram caucasoides. A triagem de mutações foi realizada pela técnica de reação em cadeia da polimerase (F508del), seguida por digestão enzimática (demais mutações). A análise clínica foi realizada utilizando as variáveis sexo, idade, etnia, manifestações pulmonares/digestivas e escore de Shwachman-Kulczycki (ESK). Resultados: Todos os pacientes apresentaram manifestações pulmonares, e 8 não apresentaram manifestações digestivas. Os resultados do ESK evidenciaram doença leve, moderada e grave, respectivamente, em 22 (42,3%), 17 (32,7%) e 13 (25,0%) pacientes. Não houve associação da mutação F508del com o grau de doença pelo ESK. Dos 140 alelos analisados, a mutação F508del foi identificada em 70 (50%). As demais mutações (G542X, G551D, R553X, R1162X e N1303K) foram identificadas em 12 (7,93%) dos alelos analisados. Em pacientes homozigotos F508del com doença grave, a OR foi de 0,124 (IC95%: 0,005-0,826). Conclusões: O diagnóstico molecular de FC foi confirmado pela identificação de apenas uma mutação (F508del) em 50% dos alelos estudados. Se considerarmos a análise das seis mutações de maior frequência na população brasileira (incluindo F508del), o diagnóstico molecular foi confirmado em 58,57% dos alelos analisados.
Palavras-chave:
Fibrose cística; Regulador de condutância transmembrana em fibrose cística; Mutação.
IntroduçãoA fibrose cística (FC; #219700) é uma doença autossômica, monogênica e recessiva, causada por mutações no gene cystic fibrosis transmembrane regulator (CFTR; #602421) na região 7q3.1.(1) Desde a identificação do gene CFTR, aproximadamente 2.000 mutações foram identificadas no mesmo.
Atualmente, o diagnóstico da FC é baseado em valores alterados de sódio e cloro no suor; contudo, outras ferramentas de diagnóstico têm sido estudadas, tais como: (a) mensuração dos valores de cloro e sódio na saliva(2); (b) indução da secreção de cloro por estímulo -adrenérgico no suor(3); (c) medidas da secreção de cloro pelo CFTR em biópsias retais(4); (d) triagem neonatal por tripsina imunorreativa(5); (e) diferença de potencial nasal(6); e (f) sequenciamento de nova geração e/ou determinação de duas mutações no gene CFTR, em seguimento ou não, da triagem neonatal.(7-9)
A gravidade da FC é dependente e modulada por fatores ambientais, genes modificadores e classes das mutações no gene CFTR.(8-17) As mutações no gene CFTR são divididas em seis classes associadas à gravidade da doença.(8,9) Mutações de classe I, II e III acarretam maior gravidade do que as de IV, V e VI.
O estudo de todas as mutações no gene CFTR não é possível na realidade brasileira e em muitos países. Nesse contexto, o presente estudo buscou a identificação das mutações de maior frequência no gene CFTR em um centro de referência. Todos os pacientes foram investigados para as mutações de maior frequência da FC - F508del (c.1521_1523delCTT; p.Phe508del; classe II); G542X (c.1624G>T; p.Gly542X; classe I); G551D (c.1652G>A; p.Gly551Asp; classe III); R553X (c.1657C>T; p.Arg553X; classe I); R1162X (c.3484C>T; p.Arg1162X; classe I); e N1303K (c.3909C>T; p.Asn1303Lys; classe II) - e os resultados foram comparados com variáveis clínico-demográficas dos pacientes.
MétodosEstudo de corte transversal. Foram incluídos 70 pacientes com FC diagnosticada por dois valores de cloro iguais ou superiores a 60 mEq/L no teste do suor.
O estudo foi aprovado pelo comitê de ética em pesquisa da instituição (Protocolo #297/2003). Todos os pacientes assinaram o termo de consentimento livre e esclarecido antes do início do estudo, e no caso de o paciente ser menor de idade, o termo foi assinado pelos pais ou responsáveis legais.
A extração de DNA foi realizada pela técnica do cloreto de lítio. A concentração de DNA utilizada para a análise foi de 50 ng/mL, avaliada em um espectrofotômetro (GE NanoVueTM; GE Healthcare Biosciences, Pittsburgh, PA, EUA).
Para a identificação das mutações no gene CFTR, o procedimento utilizado está descrito nas Tabelas 1 e 2, incluindo os iniciadores, tamanhos dos fragmentos, enzimas de restrição (segundo recomendações do fabricante) e o fragmento que representa a presença da mutação.
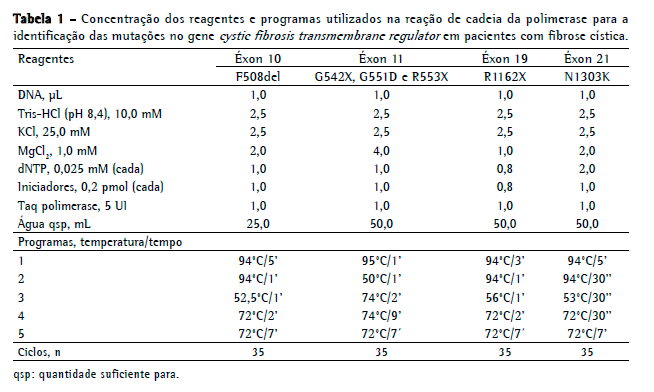

A técnica utilizada para a identificação das mutações analisadas (G542X, G551D, R553X, R1162X e N1303K) foi a digestão enzimática, exceto para a F508del, que é a deleção de três pares de base no éxon 10. No éxon 11 (G542X, G551D e R553X), quando detectada a presença de mutações, uma segunda digestão enzimática foi realizada para diferenciar as mutações G551D e R553X.
A caracterização clínica dos pacientes foi realizada por dois pesquisadores do estudo, devidamente capacitados para a análise. As variáveis utilizadas no estudo foram: sexo (masculino/feminino), idade (< 10, 11-20 e > 21 anos), etnia (caucasoides e não caucasoides), manifestações clínicas de comprometimento pulmonar (determinada por cultura de secreções do trato respiratório, espirometria e mensuração da saturação de oxigênio transcutânea), manifestações digestivas (balanço de gordura nas fezes e avaliação da elastase fecal) e escore de Shwachman-Kulczycki (ESK).
O ESK avalia a nutrição, atividade geral, exame físico e alterações radiográficas. Cada item avaliado foi pontuado para atingir o máximo de 25 pontos. Quanto menor a pontuação, pior o quadro clínico. O escore é graduado em excelente (86-100), bom (71-85), médio (56-70), moderado (41-55) e grave (40 ou menos), conforme o número total de pontos.(18) Definiu-se como doença de grau leve o escore do ESK excelente e bom; de grau moderado, o escore médio; e de grau grave, os escores moderado e grave. A categorização dos escores foi realizada para se obter um número menor de grupos para permitir a análise estatística dos dados.
A análise estatística foi realizada pelo programa IBM SPSS Statistics, versão 21.0 para Windows (IBM Corp., Armonk, NY, EUA).
Para as mutações e descrição das variáveis clínico-demográficas categóricas (sexo, etnia e presença de manifestação pulmonar/digestiva) foi utilizado o valor absoluto e percentual. Para a idade, foi utilizado à média e o desvio-padrão.
A comparação da presença das mutações com as variáveis clínico-demográficas dos pacientes foi realizada pelos testes do qui-quadrado e exato de Fisher, com o cálculo de OR, com o intuito de comparar a gravidade clínica em relação à identificação das mutações no gene CFTR incluídas no estudo.
ResultadosOs pacientes incluídos tiveram uma média de idade de 12,38 ± 9.00 anos (variação, 2-49 anos). A distribuição dos pacientes segundo as faixas etárias em estudo foram de 57,14%, 27,14% e 15,72%, respectivamente, nas faixas < 10 anos, 11-20 anos e > 21 anos.
Foram incluídos 36 pacientes do sexo feminino (51,43%). Houve distribuição uniforme em relação ao sexo.
A origem caucasoide foi observada em 94,29% dos pacientes. Todos os pacientes apresentaram manifestação respiratória, e apenas 8 (11,43%), sendo 2 deles heterozigotos compostos para a F508del e os demais sem a mutação (OR = 0,111; IC95%: 0,014-0,581), não apresentaram manifestação digestiva (Tabela 3).

A descrição dos genótipos e da frequência alélica para as mutações identificadas está apresentada na Tabela 4. Nos 52 pacientes com genótipos que continham ao menos uma das mutações estudadas, 22 (42,3%), 17 (32,7%) e 13 (25,0%), respectivamente, foram classificados como tendo doença leve, moderada e grave segundo os resultados do ESK. Não houve associação da mutação F508del com a gravidade segundo o ESK; apenas pacientes homozigotos F508del apresentaram OR de 0,124 (IC95%: 0,005-0,826) para a forma grave (Tabela 5).


Dos 140 alelos analisados, a mutação F508del foi identificada em 70 (50%). Para a frequência genotípica, a mutação F508del em homozigose ocorreu em 30% dos pacientes.
DiscussãoOs valores de distribuição das idades dos pacientes em nosso estudo foram semelhantes aos de Maróstica et al.,(19) sendo que, em 61 pacientes, a idade variou de quatro meses a 17 anos, e 73,77% dos pacientes eram menores de 10 anos. Essa distribuição deve refletir a presença de mutações das classes I-III, mais frequentemente identificadas nos centros pediátricos de FC em detrimento de maior prevalência de mutações das classes IV-VI nos centros de FC de adultos.
Como a FC é uma doença genética autossômica recessiva, é de se esperar um equilíbrio na prevalência entre os sexos, como foi por nós observado. Alguns autores têm descrito um discreto predomínio do sexo masculino, fato relatado por Streit et al.(20) e Maróstica et al.,(19) respectivamente, com valores de 61% e 62,3% para o sexo masculino. Uma hipótese é a maior deterioração da função pulmonar observada nas meninas durante a puberdade.
A prevalência de caucasoides está de acordo com os dados da literatura, que demonstram uma baixa incidência de FC entre não caucasoides. No Brasil, estudos realizados no estado do Rio Grande do Sul (predominantemente caucasoide)(19) e no estado da Bahia (predominantemente negroide)(21) evidenciaram, respectivamente, 100% e 28,7% de caucasóides com FC, demonstrando que a prevalência de FC pode estar associada com características étnicas.
Embora o diagnóstico molecular da FC seja complexo devido à heterogeneidade molecular do gene CFTR, a análise pontual de algumas mutações é necessária. Um estudo envolvendo a mutação F508del como primeiro passo para a identificação molecular de mutações no gene CFTR foi realizado por nosso grupo.(7) Dos 140 alelos analisados, a mutação F508del foi identificada em 70 (50%). A elevada prevalência da F508del pode ser observada também em estudos realizados no Rio Grande do Sul,(20) São Paulo,(22,23) Rio de Janeiro,(24) e Pará,(25) nos quais, respectivamente, a análise de 154, 116, 148, 108 e 66 alelos, mostrou a frequência de 48,7% (2 = 0,05; p = 0,82), 47% (2 = 0,17; p = 0,68), 44,45% (2 = 0,75; p = 0,38), 25,68% (2 = 18,16; p < 0,01) e 22,7% (2 = 13,77; p = 0,01). Vale ressaltar que todos esses estudos são brasileiros.
As maiores prevalências de homozigoze e heterozigoze para a mutação F508del, comparadas aos resultados em outros estados do Brasil, reforça, em nosso meio, a importância da triagem da mutação F508del.(7) Enquanto, em nosso estudo, a frequência genotípica para a mutação F508del em homozigoze ocorreu em 30% dos pacientes, no Rio Grande do Sul,(20) São Paulo(23) e Rio de Janeiro(24) e essa ocorreu, respectivamente, em 31,2%, 21,3% e 10,81% dos pacientes. Por outro lado, em nosso estudo, a heterozigose ocorreu em 40,01%, que é um valor mais próximo do que os encontrados nos mesmos estudos acima citados (28,6%, 46,3% e 22,97%, respectivamente).(20,23,24)
A prevalência da mutação G542X foi de 4,29% em nosso estudo, valor semelhante aos encontrados em outros estudos no Brasil, de 3,2% (2= 6,22; p = 0,63),(20) 2,7% (2= 0,54; p = 0,46)(24) e 5,5 (2 = 11,91; p < 0,01).(26) A mutação R553X foi identificada em 0,71% dos alelos analisados. Nos estudos de Streit et al.(20) e Raskin et al.,(26) houve frequência de 0,7% (2 = 0,01; p = 0,93) e 0,8% (2 = 4,31; p = 0,037), respectivamente. A mutação R1162X foi detectada em 2,4% dos alelos. Embora as mutações de classe I (G542X, R553X e R1162X) tenham baixa prevalência no Brasil (menos que 6% dos alelos pesquisados), vale ressaltar que estudos em modelos animais e humanos têm sido promissores para novas drogas corretoras da função da CFTR para essas mutações, entre elas, PTC124.(27)
Ao contrário da mutação F508del, também de classe II, com alta prevalência, a N1303K apresentou baixa prevalência (1,43% dos alelos analisados). Outros estudos brasileiros não analisaram essa mutação.(20,26) Para essa classe de mutação, drogas corretoras e potenciadoras da função da CFTR têm sido pesquisadas, tais como VX-770 e VX-809.(28,29)
A mutação G551D não foi detectada na amostra avaliada, sendo apenas identificada por Raskin et al.(26) em um alelo analisado (1,8%; 2 = 2,51; p = 0,11). Para essa mutação, está disponível no mercado a droga VX-770, conhecida como kalydeco (Vertex®).(28)
Em nossa casuística de 70 pacientes com FC, 8 pacientes não apresentaram alteração digestiva. Desses, nenhum era homozigoto F508del e, em 5, não foi detectada nenhuma das mutações estudadas.
Embora a relação da mutação F508del e a gravidade da doença seja conhecida na associação entre essa mutação e resultados do ESK, 1,92% dos indivíduos homozigotos para a mutação F508del apresentava doença grave, enquanto, naqueles com duas mutações não diagnosticadas, 23,08% dos indivíduos apresentava doença grave segundo o ESK. Algumas hipóteses podem ser formuladas para a menor frequência de doença grave pelo ESK em pacientes homozigotos F508del, tais como a modulação gênica,(8-17) a maior mortalidade precoce desses pacientes e a baixa adesão ao manejo ou ao tratamento da doença.
Concluímos que a análise das seis mutações do gene CFTR permitiu a identificação de 58,57% dos 140 alelos. Contudo, do total analisado, 50% dos alelos eram da mutação F508del, que deve ser o padrão inicial para a identificação de mutações no gene CFTR, tendo como base a recente implantação do teste de triagem neonatal em nosso estado. A presença de resultados ESK indicando doença leve para a maioria dos pacientes, mesmo naqueles com mutações de classe II em forma homozigota, pode traduzir a baixa idade dos pacientes que ainda não apresentam gravidade clínica associada à progressão da doença, bem como a baixa adesão dos pacientes de maior gravidade ao tratamento da doença.
Referências1. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066-73. Erratum in: Science 1989;245(4925):1437. http://dx.doi.org/10.1126/science.2475911 PMid:2475911
2. Gonçalves AC, Marson FA, Mendonça RM, Ribeiro JD, Ribeiro AF, Paschoal IA, et al. Saliva as a potential tool for cystic fibrosis diagnosis. Diagn Pathol. 2013;8:46. http://dx.doi.org/10.1186/1746-1596-8-46 PMid:23510227 PMCid:PMC3621375
3. Quinton P, Molyneux L, Ip W, Dupuis A, Avolio J, Tullis E, et al. -adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am J Respir Crit Care Med. 2012;186(8):732-9. http://dx.doi.org/10.1164/rccm.201205-0922OC PMid:22859523
4. Sousa M, Servidoni MF, Vinagre AM, Ramalho AS, Bonadia LC, Felício V, et al. Measurements of CFTR-mediated Cl-secretion in human rectal biopsies constitute a robust biomarker for cystic fibrosis diagnosis and prognosis. PLoS One. 2012;7(10):e47708. http://dx.doi.org/10.1371/journal.pone.0047708 PMid:23082198 PMCid:PMC3474728
5. Wagener JS, Zemanick ET, Sontag MK. Newborn screening for cystic fibrosis. Curr Opin Pediatr. 2012;24(3):329-35. http://dx.doi.org/10.1097/MOP.0b013e328353489a PMid:22491493
6. Valiulis A, Skurvydienė I, Misevičienė V, Kasnauskienė J, Vaidelienė L, Utkus A. Relevance of nasal potential difference in diagnosis of cystic fibrosis among children. Medicina (Kaunas). 2013;49(4):185-90.
7. Marson FA, Bertuzzo CS, Ribeiro MÂ, Ribeiro AF, Ribeiro JD. Screening for F508del as a first step in the molecular diagnosis of cystic fibrosis. J Bras Pneumol. 2013;39(3):306-16. PMid:23857699
8. Knowles MR, Drumm M. The influence of genetics on cystic fibrosis phenotypes. Cold Spring Harb Perspect Med. 2012;2(12):a009548. http://dx.doi.org/10.1101/cshperspect.a009548 PMid:23209180
9. Drumm ML, Ziady AG, Davis PB. Genetic variation and clinical heterogeneity in cystic fibrosis. Annu Rev Pathol. 2012;7:267-82. http://dx.doi.org/10.1146/annurev-pathol-011811-120900 PMid:22017581
10. Faria EJ, Faria IC, Ribeiro JD, Ribeiro AF, Hessel G, Bertuzzo CS Association of MBL2, TGF-beta1 and CD14 gene polymorphisms with lung disease severity in cystic fibrosis. J Bras Pneumol. 2009;35(4):334-42. http://dx.doi.org/10.1590/S1806-37132009000400007 PMid:19466271
11. Lima CS, Ortega MM, Marson FA, Zulli R, Ribeiro AF, Bertuzzo CS. Cystic fibrosis transmembrane conductance regulator gene mutations and glutathione S-transferase null genotypes in cystic fibrosis patients in Brazil. J Bras Pneumol. 2012;38(1):50-6. http://dx.doi.org/10.1590/S1806-
37132012000100008 PMid:22407040
12. Marson FA, Bertuzzo CS, Ribeiro AF, Ribeiro JD. Polymorphisms in ADRB2 gene can modulate the response to bronchodilators and the severity of cystic fibrosis. BMC Pulm Med. 2012;12:50. http://dx.doi.org/10.1186/1471-2466-12-50 PMid:22950544 PMCid:PMC3558405
13. Marson FA, Bertuzzo CS, Hortencio TD, Ribeiro JD, Bonadia LC, Ribeiro AF. The ACE gene D/I polymorphism as a modulator of severity of cystic fibrosis. BMC Pulm Med. 2012;12:41. http://dx.doi.org/10.1186/1471-2466-12-41 PMid:22874010 PMCid:PMC3460779
14. Marson FA, Marcelino AR, Ribeiro AF, Ribeiro JD, Bertuzzo CS. COX-2 gene polymorphisms: genetic determinants of cystic fibrosis comorbidities. Int J Genet. 2013;5(1):132-8.
15. Marson FAL, Rezende LM, Furgeri DT, Ribeiro AF, Ribeiro JD, Bertuzzo CS. ADRA2A is a cystic fibrosis modifier gene. Int J Genet. 2013;5(1)1:125-31.
16. de Lima Marson FA, Bertuzzo CS, Secolin R, Ribeiro AF, Ribeiro JD. Genetic interaction of GSH metabolic pathway genes in cystic fibrosis. BMC Med Genet. 2013;14:60. http://dx.doi.org/10.1186/1471-2350-14-60 PMid:23758905 PMCid:PMC3685592
17. Furgeri DT, Marson FA, Ribeiro AF, Bertuzzo CS. Association between the IVS4G>T mutation in the TCF7L2 gene and susceptibility to diabetes in cystic fibrosis patients. BMC Res Notes. 2012;5:561. http://dx.doi.org/10.1186/1756-0500-5-561 PMid:23050589 PMCid:PMC3519805
18. Santos CI, Ribeiro JD, Ribeiro AF, Hessel G. Critical analysis of scoring systems used in the assessment of cystic fibrosis severity: state of the art. J Bras Pneumol. 2004;30(3):286-98.
19. Maróstica PJ, Raskin S, Abreu-e-Silva FA. Analysis of the delta F508 mutation in a Brazilian cystic fibrosis population: comparison of pulmonary status of homozygotes with other patients. Braz J Med Biol Res. 1998;31(4):529-32. http://dx.doi.org/10.1590/S0100-879X1998000400009 PMid:9698805
20. Streit C, Burlamaque-Neto AC, de Abreu e Silva F, Giugliani R, Saraiva Pereira ML. CFTR gene: molecular analysis in patients from South Brazil. Mol Genet Metab. 2003;78(4):259-64. http://dx.doi.org/10.1016/S1096-7192(03)00033-7
21. Santana MA, Matos E, do Socorro Fontoura M, Franco R, Barreto D, Lemos AC. Prevalence of pathogens in cystic fibrosis patients in Bahia, Brazil. Braz J Infect Dis. 2003;7(1):69-72. http://dx.doi.org/10.1590/S1413-86702003000100008 PMid:12807693
22. Raskin S, Phillips JA 3rd, Krishnamani MR, Vnencak-Jones C, Parker RA, Rozov T, et al. DNA analysis of cystic fibrosis in Brazil by direct PCR amplification from Guthrie cards. Am J Med Genet. 1993;46(6):665-9. http://dx.doi.org/10.1002/ajmg.1320460612 PMid:8362909
23. Cabello GM, Cabello EH Jr JL, Fernande O, Harris A. The 3120 +1G-->A splicing mutation in CFTR is common in Brazilian cystic fibrosis patients. Hum Biol. 2001;73(3):403-9. http://dx.doi.org/10.1353/hub.2001.0031 PMid:11459421
24. Okay TS, Oliveira WP, Raiz-Júnior R, Rodrigues JC, Del Negro GM. Frequency of the deltaF508 mutation in 108 cystic fibrosis patients in Sao Paulo: comparison with reported Brazilian data. Clinics (Sao Paulo). 2005;60(2):131-4. http://dx.doi.org/10.1590/S1807-59322005000200009
25. Araújo FG, Novaes FC, Santos NP, Martins VC, Souza SM, Santos SE, et al. Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil. Braz J Med Biol Res. 2005;38(1):11-5. http://dx.doi.org/10.1590/S0100-879X2005000100003 PMid:15665983
26. Raskin S, Phillips JA, Kaplan G, McClure M, Vnencak-Jones C, Rozov T, et al. Geographic heterogeneity of 4 common worldwide cystic fibrosis non-DF508 mutations in Brazil. Hum Biol. 1999;71(1):111-21. PMid:9972102
27. Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci U S A. 2008;105(6):2064-9. http://dx.doi.org/10.1073/pnas.0711795105 PMid:18272502 PMCid:PMC2538881
28. Leonard A, Leal T, Lebecque P. Mucoviscidosis: CFTR mutation-specific therapy: a ray of sunshine in a cloudy sky [Article in French]. Arch Pediatr. 2013;20(1):63-73. http://dx.doi.org/10.1016/j.arcped.2012.10.018 PMid:23199563
29. Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordo-ez CL, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142(3):718-24. http://dx.doi.org/10.1378/chest.11-2672 PMid:22383668 PMCid:PMC3435140
* Trabalho realizado no Departamento de Genética Médica e Departamento de Pediatria, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas (SP) Brasil.
Endereço para correspondência: Fernando Augusto de Lima Marson. Universidade Estadual de Campinas, Faculdade Ciências Médicas, Departamento de Genética Médica. Rua Tessália Vieira de Camargo, 126, Cidade Universitária "Zeferino Vaz", CEP 13083-887, Campinas, SP, Brasil.
Tel. 55 19 3521-8902. Fax: 55 19 3521-8909. E-mail: fernandolimamarson@hotmail.com
Apoio financeiro: Nenhum.
Recebido para publicação em 15/5/2013. Aprovado, após revisão, em 17/9/2013.
Sobre os autoresCyntia Arivabeni de Araújo Correia Coutinho
Pesquisadora. Departamento de Genética Médica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas (SP) Brasil.
Fernando Augusto de Lima Marson
Doutorando. Departamento de Genética Médica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas (SP) Brasil.
Antônio Fernando Ribeiro
Professor. Departamento de Pediatria, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas (SP) Brasil.
José Dirceu Ribeiro
Professor Titular. Departamento de Pediatria, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas (SP) Brasil.
Carmen Silvia Bertuzzo
Professora. Departamento de Genética Médica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas (SP) Brasil.












 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket