ABSTRACT
Background: Structural alterations to the pulmonary circulation characterize the vascular remodeling process and are likely correlated with local variations in flow and ischemia.
Objective: To define the histological alterations to the pulmonary circulation seen after experimentally-induced ischemia of the pulmonary artery and to correlate those alterations with known patterns of blood redistribution and vascular remodeling.
Method: Wistar rats (n = 48) were randomized into two groups with ligation of the pulmonary artery and without (controls) and were sacrificed on post-ischemia days 1, 7, 30 and 60. Lungs were removed and inspected for signs of parenchymal injury. External diameters, as well as wall thicknesses in the pulmonary, alveolar and bronchial end arterioles, were measured. Internal diameter and wall thickness percentage were calculated.
Results: Infarction, necrosis and hemorrhage occurred only in ischemic lungs. In nonischemic lungs, there was a sustained increase in the internal and external arteriolar diameters, with an initial reduction in wall thickness on day 1, and day-60 values were similar to those seen in controls. In ischemic lungs, there was a transitory reduction in the internal and external diameters of the pulmonary and bronchial end arterioles, together with an initial, equally transitory, increase in their wall thickness. The alveolar arterioles presented sustained and progressive increases in external diameter and wall thickness, with concomitant reductions in internal diameter.
Conclusion: This model mimics distal arterial disease in patients with chronic pulmonary thromboembolism. The vascular response in nonischemic lungs was consistent with a pattern of flow remodeling, whereas that seen in ischemic lungs was more consistent with flow and ischemia. In the pulmonary and bronchial end arterioles, the response was transitory, in contrast to the sustained and progressive response seen in the alveolar arterioles, which was probably caused by delayed local flow.
Keywords:
Key-words: Pulmonary embolism. Pulmonary circulation. Vascular remodelling. Pulmonary artery.
RESUMO
Introdução: Alterações estruturais da circulação pulmonar traduzem processo de remodelação vascular e têm relação provável com variações locais de fluxo e isquemia.
Objetivo: Definir as alterações histológicas na circulação pulmonar após obstrução experimental da artéria pulmonar. Correlacioná-las com os padrões de redistribuição sangüínea e remodelação vascular.
Método: Foram submetidos à toracotomia esquerda 48 ratos Wistar, alocados aleatoriamente em dois grupos, com ligadura da artéria pulmonar e controle, e sacrificados com 1, 7, 30 e 60 dias. Nos pulmões retirados avaliou-se presença de sinais de injúria no parênquima e mensurou-se diâmetro externo e espessura da parede das arteríolas de bronquíolos terminais, respiratórios e alveolares. Diâmetro interno e porcentagem de espessura da parede foram calculados.
Resultados: Só ocorreu infarto, necrose e hemorragia no pulmão isquêmico. No não isquêmico houve aumento mantido dos diâmetros externo e interno das arteríolas, com redução inicial da espessura no 1o dia e valores semelhantes aos do grupo controle no 60o dia. No pulmão isquêmico houve redução transitória nos diâmetros externo e interno das arteríolas de bronquíolos terminais e respiratórios, com aumento, inicial e transitório, na sua espessura. As arteríolas alveolares apresentaram aumento do diâmetro externo e espessura da parede, com redução do diâmetro interno, mantida e progressiva.
Conclusão: Este modelo reproduz arteriopatia distal em pacientes com tromboembolismo pulmonar crônico. A resposta vascular no pulmão não isquêmico é compatível com padrão de remodelação de hiperfluxo; a no pulmão isquêmico com hipofluxo e isquemia. Nas arteríolas de bronquíolos terminais e respiratórios a resposta foi transitória. Nas alveolares foi progressiva e mantida, pela provável ocorrência tardia de hiperfluxo local.
Palavras-chave:
Descritores: Embolia pulmonar. Circulação pulmonar. Remodelação vascular. Artéria pulmonar.
Introduction
The occurrence of chronic pulmonary embolism is attributed to frequently recurring episodes of acute thromboembolism, in which there is nonlytic organization of the thrombi(1). It is believed that persistence of this obstruction results in elevated blood pressure levels and, consequently, high mortality rates(2,3). Nevertheless, patients with chronic pulmonary thromboembolism may remain asymptomatic for several years before presenting worsening of hemodynamic symptoms. Evidence shows that alterations in the pulmonary microvasculature (arterial disease of the small blood vessels) may be, in most cases, the determinant factor in the progressive worsening of these patients.
Patients with chronic pulmonary thromboembolism and distal arterial disease (hypertrophy of the middle layer, alterations to the inner layer, plexiform lesions and organized thrombi) present worse evolution after pulmonary endarterectomy (rethrombosis and residual tricuspid insufficiency, as well as elevated pulmonary arterial pressure and vascular resistance), when compared to patients without concomitant distal arterial disease(4).
It is controversial whether the arterial disease seen in chronic pulmonary thromboembolism is due to unspecified effects of persistent pulmonary hypertension(5), which some authors suggest is the final step in the vascular remodeling phenomenon(6).
By measuring the pulmonary artery response in an experimental model of pulmonary postobstructive vasculopathy, it is possible to establish remodeling patterns over time(7). Etiological factors possibly involved in chronic pulmonary thromboembolism, such as the occurrence of anastomoses between the bronchial and the pulmonary systems(8), ischemic endothelial injury(8) and variations in blood flow through the arterial system(9), could be related to these response patterns.
The objective of this study was to define the histological alterations to the pulmonary circulation, in both ischemic and nonischemic areas, seen after experimentally-induced ischemia of the pulmonary artery and to correlate those alterations with known patterns of blood redistribution and vascular remodeling, pre-established in the literature.
Methods
This study was carried out in the Medical Investigation Laboratories LIM-61 (Thoracic Surgery Research) and LIM-20 (Experimental Therapy
Research), and in the Pathological Anatomy Department of the Instituto do Coração of the Faculdade de Medicina da Universidade de São Paulo. The experiment followed the norms established in the Guide for the Care and Use of Laboratory Animals(10), the Brazilian Legislation Ethical Principles of Animal Experimentation(11) and the Brazilian College of Experimentation(12).
Healthy adult male Wistar rats (n = 48), weighing 200-400 g, were used. In the preoperative period, the animals were randomized into two initial groups, designated ligation (n = 28), and control (n = 20). In the postoperative period, the animals in both groups were randomized into four sub-groups according to the time of sacrifice (day 1, 7, 30 or 60).
The animals were anesthetized via inhalation with a mixture of 2% isoflurane (Isothane-Zeneca) under O2 at 100%. Tidal volume was adjusted to 10 ml/kg and respiratory frequency was maintained at 80 cycles per minute.
All of the animals underwent left thoracotomy and dissection of the left pulmonary artery. In the ligation group, the artery was occluded with a surgical clip (Premium Surgiclip M-11.5; Autosuture, Norwalk, CT, USA), whereas no clips were used in control group animals. Postoperatively, the animals were sacrificed by exsanguination at specific times (days 1, 7, 30 and 60). The lungs were removed en bloc and fixed by tracheal perfusion with 4% paraformaldehyde in 0.1 phosphate buffer (pH 7.3) until pleural tension was obtained, after which the trachea was clamped(13-15).
The histological sections were stained using hematoxylin and eosin (H&E) and Verhoeff's elastin stain for elastic fibers. The laminae were coded (veiled) and evaluated by two independent observers using an optical microscope (Q500 YW; Leica, Cambridge, UK), with 40x magnification, attached to an image analyzer (Quantimet Q500 YW; Leica). The histological sections, from the right and left lungs of both ligation group and control group animals, were analyzed quantitatively and qualitatively.
In the qualitative analysis, we studied the overall evidence of cellular injury (infarction, necrosis and hemorrhage), intimal proliferation and obliteration of the arterial lumen. These alterations are expressed in frequency (percentage) and intensity (1+, 2+ and 3+) (Table 1).
The measurements taken in the quantitative analysis were established through a computer program for analysis and quantification of images (Quantimet; Leica). This study analyzed the pulmonary arterioles in the acinar and pre-acinar spaces, when they clearly presented external and internal elastic laminae, separated or not by muscle(10,15,16). The external diameter(17) and the thickness of the middle layer(17) of the arterioles evaluated were measured, which allowed the percentage of thickness, as well as the internal diameter, to be calculated. The percentage of mid-layer thickness was calculated using the formula(17):
2 x thickness of the middle layer x 100
External diameter
Internal diameter was calculated using the formula(18):
Internal diameter = External diameter - (2 X thickness of the middle layer)
The measurements obtained - external and internal diameters, thickness of the mid-layer and percentage of thickness - were stratified by location of the arteriole in the pulmonary parenchyma: bronchial end arterioles, pulmonary arterioles and alveolar arterioles (alveolar duct and alveolar septum)(19). These measurements allowed for comparisons between the lungs of control group and ligation group animals, throughout the postoperative period (days 1, 7, 30 and 60).
In the statistical analysis, all variables were initially analyzed descriptively, involving observation of minimum and maximum values, as well as calculation of means and standard deviation. For qualitative variables, absolute and relative frequencies were calculated and expressed as percentages. In order to test the hypothesis of equality of means, three-factor analysis of variance(20) was used. The three factors were group (control group or ligation group), postoperative period time point (day 1, 7, 30 or 60) and lung (right or left). Multiple comparisons were made by drawing contrasts among the averages based on the least squares. The level of significance used for the tests was 5%.
Results
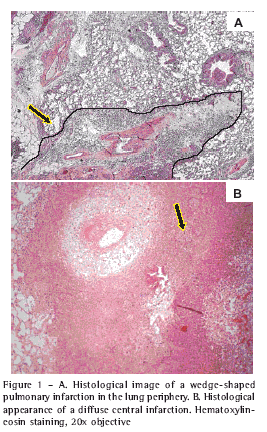
On postoperative days 1 and 7, signs of cellular injury (infarction and necrosis) were found exclusively in the ischemic lungs. The affected area was less than 50% of the surface of the histological section, varying between wedge-shaped and diffuse (Figures 1A and 1B). Hemorrhage was always associated with the area of infarction and there was no hemorrhage without infarction. From postoperative day 7 onward, intimal proliferation and obliteration of the lumen occurred (Figures 2A and 2B), also exclusively in ischemic lungs and presenting temporal variations in frequency, number of arteries affected and intensity over the course of the evolution (Table 1).
A total of 3068 arterioles were measured in both ischemic and nonischemic lungs (1105 from the control group and 1963 from the ligation group). The results express the data obtained from the ligation group in comparison to those obtained from the control group. The percentage of thickness defines the vascular response since it reflects the actual impact of variations in wall thickness on variations in external diameter and consequently on the internal diameter and arteriolar lumen.

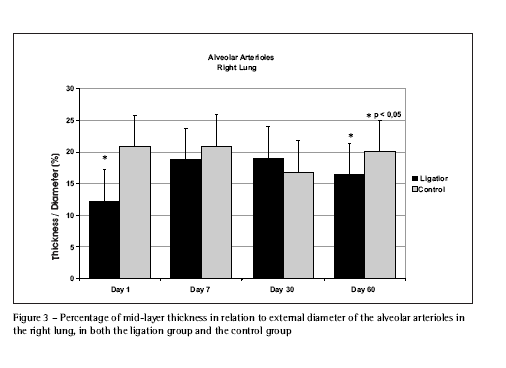
In nonischemic lungs, there was a sustained increase in the internal and external diameters of the evaluated arterioles from day 1 through day 60, more evident in the more distal (alveolar) arterioles. There was a significant reduction in the percentage of wall thickness in relation to external diameter on day 1 (Figure 3). On day 7, the percentage of thickness in the pulmonary and bronchial end arterioles presented a significant increase due to the increase in their wall thickness. Over time, there was a reduction in the percentage of thickness, which reflected the reduced wall thickness in the pulmonary and bronchial end arterioles, especially when the external diameters increased. By day 60, the arterioles showed a thickness percentage similar to the control group. The alveolar arterioles, despite still presenting increased wall thickness, presented a significant day-60 reduction in the percentage of thickness in relation to the control group (Figure 3).
As a vascular response, diameters (internal and external) of the more proximal arterioles (bronchial end and pulmonary) were reduced in the ischemic lungs at all time points except day 7, on which there was an isolated and transitory increase in these diameters in the bronchial and pulmonary arterioles. In addition, wall thickness and thickness percentage in the pulmonary and bronchial end arterioles presented an initial decrease on day 1, with transitory increases on days 7 and 30, and were similar to those seen in the control group lungs on day 60.
External diameters of the distal (alveolar) arterioles were increased at all time points, with a progressive reduction in internal diameter after day 7, paralleling the progressive increase in the thickness percentage (Figure 4).
Discussion
This experimental model employed adult Wistar rats, based on previous studies in which this species was used to establish vascular patterns and responses propitious to research into human diseases such as pulmonary hypertension and embolism(9,21,22-25).
The authors of some previous experimental studies(26-28) have committed a methodological error in utilizing the right lung as a control and submitting the left lung to ligation, ignoring the relationship between the vascular responses of the two lungs. In our study, the comparison with a true control group allowed us to identify not only the vascular response directly related to the ligation group, but also confounding factors, such as variations in pulmonary volume caused by thoracotomy, which produce alterations to the vascular structure. Such factors may constitute limitations of this study model. On day 30, for example, unexpected dimensions were found in the control group in comparison to the ligation group, making it difficult to understand the mechanisms of the vascular response.
Another potential limitation of our study is the fact that hemodynamic conditions were not monitored. We were therefore unable to correlate levels of pulmonary arterial pressure with histological findings. However, this limitation is mitigated by pre-established experimental and clinical knowledge of the patterns of vascular remodeling present under specific hemodynamic conditions(29).
Pulmonary morphological studies after the central ligation of the pulmonary artery differ in the way they obtain evidence of cellular injury, such as hemorrhage, necrosis and infarction, within the first 7 days(25,30-33). Variations in the response to arterial obstruction are attributed to adaptive changes in the bronchial circulation and bronchopulmonary collateral circulation, which, within a few days, replenish the ischemic parenchyma(24,34). The results of this study provide evidence of ischemic cellular injury (infarction, necrosis, hemorrhage) in the initial periods (on days 1 and 7), indicating that the bronchial circulation does not always nurture the parenchyma, and that ischemia may be the factor that triggers the subsequent histological alterations(31).
In chronic pulmonary hypertension, as in chronic pulmonary thromboembolism, the presence of intimal proliferation and arterial obliteration reflect advanced stages of arteriopathy(19), in which the intimal proliferation precedes the obliteration of the arteriolar lumen(2,5). Similar to an example in the literature, the arterioles found to be most affected in our study were the alveolar arterioles(35). Over time, arterioles presenting intimal proliferation reduced in number and were probably replaced by obstructed arterioles(2,5,19,35) (Table 1).
Previous studies have established patterns of vascular remodeling (increase and reduction in the diameter or wall thickness of the arterioles) mainly in response to variations (reductions or increases) in blood flow, whether accompanied by ischemia or not. Although our results do not include such findings, we used this previous experience in forming our conclusions, comparing them with the dimensional variations observed in the arterioles, in various locations and over time. Some of these aspects already established in literature must therefore be stressed so that we can better understand our findings: pulmonary arterioles submitted to increased blood flow (without ischemic lesion), after the initial increase in diameter and reduction of thickness, present transitory hypertrophy of the wall(36,39); in acute experimental and clinical pulmonary embolism, the phenomenon of ischemic endothelial injury is seen(40); ischemic endothelial lesion produces hypertrophy in the middle layer(41); in reduced blood flow, the vascular response of reducing diameter is inhibited if the endothelium is removed(42); in response to the obstruction of the pulmonary artery, the bronchial arteries respond with hypertrophy and proliferation, which initiates in the center and propagates to the periphery, and the more centrally located (pre-acinar) pulmonary arterioles are more rapidly supplied with blood than those in the intra-acinar space(43); in addition to the collateral circulation, pre-capillary anastomoses appear between the pulmonary and bronchial circulations, which open at the level of the respiratory bronchioles(44), producing local alterations of intimal proliferation(45); the combination of ischemic endothelial injury and these local hemodynamic variations produces a vascular remodeling response in the small blood vessels, resulting in intimal proliferation, a condition not seen in models of isolated injury(37).
The increased diameters and significantly lower thickness percentages observed in the arterioles of the nonischemic (right) lungs on day 1 (produced by the acute distribution of blood flow to that lung) are compatible with the pattern of increased blood flow remodeling(38) (Figure 3). On day 7, in addition to the increased external diameter, the effect of the increased blood flow on the endothelium should result in an increase in the percentage of pulmonary and bronchial end arteriole thickness(39).
Over time, the reduction in thickness percentage reflects the reduction in pulmonary and bronchial end arteriole wall thickness, more so in conjunction with greater external diameters. On day 60, the wall thickness of these arterioles were similar to that seen in those of the control group, confirming the transitory effect of the force that the increased blood flow exerts on the endothelium(36,37). On day 60, the alveolar arterioles, despite the persistence of their greater wall thickness, showed a significant reduction in the percentage of thickness in comparison to the control group, suggesting an ever-increasing predominance of arterial dilatation in relation to the transitory hypertrophy of the wall (Figure 3).
In our study, the vascular response in the left lung (submitted to ligation) varied according to the location of the arterioles (intra-acinar or pre-acinar) and according to the duration of the ligation. Therefore, the dilatation of the alveolar arterioles seen on day 1 of the ligation was compatible with a vascular response to the ischemic endothelial injury(40), in which the usual response to increased blood flow (reduced diameters) would have been inhibited(42). This fact, together with the reduction in wall thickness, led to a reduced percentage of thickness in relation to the control group (Figure 4). The lesser impact of ischemia on the pulmonary and bronchial end arterioles, maintaining the reduction in diameters through reduced blood flow, is likely due to the greater efficacy of the blood flow restoration mechanisms of the bronchial circulation in the preacinar regions(43). The vascular response seen from day 7 onward may be explained in part by the mechanisms that were active on day 1, such as endothelial lesion and reduced blood flow, and in part by the precapillary anastomoses in the pulmonary and bronchial circulations established during this ligation period(45).
The respiratory bronchiole arterioles present isolated increases in their internal and external diameters on day 7, which may be related to the precapillary anastomoses that open in the region of the respiratory bronchioles at that approximate time(43). This increase in diameters is accompanied by an increase in the percentage of thickness, both in the bronchial end arterioles and in the pulmonary arterioles, although less pronounced in the latter on day 7 due to the isolated elevation of the external diameter. In the pulmonary and bronchial end arterioles, the response pattern is determined by acute but transitory mechanisms, as evidenced by the fact that, on day 60, both present dimensions similar to those seen in the control group(44).
In contrast, the mechanisms responsible for the response in the alveolar arterioles established a permanent and progressive pattern. Therefore, from day 7 of the ligation, the thickness percentage in these arterioles increased progressively, followed by progressive reduction in their internal diameters (Figure 4). The external diameters of these arterioles remained increased up to day 60. Endothelial injury per se does not result in permanent and progressive structural alterations to the arterial wall unless accompanied by modifications to the local hemodynamic parameters(37).
Hence, it was expected that, if the response of the alveolar arterioles were exclusively dependant on the ischemic endothelial lesion, even when more intense, the day-60 thickness percentages for these arterioles would be, as seen in the pulmonary and bronchial end arterioles, similar to those seen in the control group. This progressive and permanent response in the intra-acinar vascular bed is likely attributable to the initial ischemic endothelial lesion and the blood flow through the precapillary anastomoses(44), together constituting a trigger mechanism for the local hemodynamic modification.
Above all, despite not using embolic mechanisms, this model of pulmonary artery ligation reproduced, after 60 days of obstruction, the distal arteriopathy identified in patients with chronic pulmonary thromboembolism, thereby suggesting that the mechanisms of ischemia and flow distribution resulting from arterial obstruction may actually be responsible for the obstruction. This arteriopathy is in its initial phase (hypertrophy of the middle layer and intimal proliferation), suggesting a still reversible pattern, which will allow the continuity of the study with the evaluation of the mechanisms and response involved in reperfusion.
As previously mentioned, we know that early intervention in chronic pulmonary embolism improves postoperative evolution of the patients, especially when they have not yet presented irreversible sequelae of a long, previously undiagnosed, process of vascular remodeling(4).
Therefore, from the standpoint of clinical reproducibility, the understanding obtained from this response might contribute to the establishment of better prophylactic treatments for arteriopathy after an acute episode, as well as improving thromboendarterectomy outcomes, either by influencing the selection of candidates, identifying predictors of postoperative evolution or guiding the development of new preoperative and intraoperative procedures, with the end result of reducing morbidity and mortality(21).
References
1. Dalen JE. Pulmonary embolism: what have we learned since Virchow? Natural history, pathophysiology, and diagnosis. Chest 2002; 122:1440-56.
2. Wagenvoort CA. Pathology of pulmonary thromboembolism. Chest 1995; 107 (1 Suppl):10S-17S.
3. Riedel M, Stanek V, Widimsky J, Prerovsky I. Long term follow-up of patients with pulmonary thromboembolism. Late prognosis and evolution of hemodynamic and respiratory data. Chest 1982; 81:151-8.
4. Thistlethwaite PA, Mo M, Madani MM, Deutsch R, Blanchard D, Kapelanski DP, et al. Operative classification of thromboembolic disease determines outcome after pulmonary endarterectomy. J Thorac Cardiovasc Surg 2002; 124:1203-11.
5. Moser KM, Bloor CM. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest 1993;103:685-92.
6. Dzau VJ, Horiuchi M. Vascular remodeling: the emerging paradigm of programmed cell death (apoptosis): the Francis B. Parker lectureship. Chest 1998; 114 (1 Suppl): 91S-9S.
7. Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med 1994; 330: 1431-8.
8. Johnson JE, Perkett EA, Meyrick B. Pulmonary veins and bronchial vessels undergo remodeling in sustained pulmonary hypertension induced by continuous air embolization into sheep. Exp Lung Res 1997; 23:459-73.
9. Tanaka Y, Schuster DP, Davis EC, Patterson GA, Botney MD. The role of vascular injury and hemodynamics in rat pulmonary artery remodeling. J Clin Invest 1996; 98:434-42.
10.Haworth SG. Development of the normal and hypertensive pulmonary vasculature. Exp Physiol 1995; 80: 843-53.
11.Guide for the care and use of laboratory animals. Institute of laboratory animal resources. National Research Council of the National Academy of Sciences. Washington, D.C. National Academy Press; 1996. p.1-35.
12.Normas para a prática didático-científica da vivissecação de animais. Lei da República Federativa do Brasil nO 6638, artigos 1-8 (08 de maio de 1979). Disponível em URL: http:// www.meusite.com.br/COBEA.
13.Princípios éticos na experimentação animal. Colégio Brasileiro de Experimentação Animal. Disponível em URL: http://www.meusite.com.br/COBEA.
14.Cabrera Fischer EI, Willshaw P, de Forteza E, Biagetti M, Altman R, Morales MC, et al. Animal model of acute pulmonary thromboembolism treated by local recirculation of streptokinase through the lung. J Thorac Cardiovasc Surg 1987; 93: 620-7.
15.Kasinski N, Ferreira C, Giannotti Filho O, Afonso JE, Lopes dos Santos M, Barcellini A. Pulmonary structural changes in thromboembolism. Experimental study in dogs. Rev Paul Med 1984; 102:7-12.
16.Meyrick B, Hislop A, Reid L. Pulmonary arteries of the normal rat: the thick walled oblique muscle segment. J Anat 1978;125: 209-21.
17.Hislop A, Reid L. Normal structure and dimensions of the pulmonary arteries in the rat. J Anat 1978; 125: 71-83.
18.Davies G, Reid L. Growth of the alveoli and pulmonary arteries in childhood. Thorax 1970; 25: 669-81.
19.Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension 1989; 13:968-72.
20.Rabinovitch M, Haworth SG, Castaneda AR, Nadas AS, Reid LM. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 1978; 58:1107-22.
21.Rosner B. Fundamentals of Biostatistics. 2nd ed. Boston: PWS Publishers; 1986.
22.Takeoka M, Sakai A, Ueda G, Ge RL, Panos RJ, Taniguchi S. Influence of hypoxia and pulmonary air embolism on lung injury in perfused rat lungs. Respiration 1996; 63:346-51.
23.Wang D, Li MH, Hsu K, Shen CY, Chen HI, Lin YC. Air embolism-induced lung injury in isolated rat lungs. J Appl Physiol 1992; 72:1235-42.
24.Weibel ER. Early stages in the development of collateral circulation to the lung in the rat. Circ Res 1960; 8:353-76.
25.Liebow AA, Hales MR, Bloomer WE, Harrison W, Lindskog GE. Studies on the lung after ligation of the pulmonary artery. II. Anatomical changes. Am J Pathol 1950; 26: 177-85.
26.Michel RP, Hakim TS. Increased resistance in postobstructive pulmonary vasculopathy: structure-function relationships. J Appl Physiol 1991; 71: 601-10.
27.Shi W, Hu F, Kassouf W, Michel RP. Altered reactivity of pulmonary vessels in postobstructive pulmonary vasculopathy. J Appl Physiol 2000; 88:17-25.
28.Shi W, Cernacek P, Hu F, Michel RP. Endothelin reactivity and receptor profile of pulmonary vessels in postobstructive pulmonary vasculopathy. Am J Physiol 1997; 273:H2558-64.
29.Johnson RL Jr, Cassidy SS, Haynes M, Reynolds RL, Schulz W. Microvascular injury distal to unilateral pulmonary artery occlusion. J Appl Physiol 1981; 51: 845-51.
30.Virchow RLK. Gesammelte abhandlungen zur wissenschaftlichen medicin. Frankfurt, Germany: Meidinger sohn u Comp, 1856; 294-6.
31. Morgan TE, Edmunds LH Jr. Pulmonary artery occlusion. II. Morphologic studies. J Appl Physiol 1967; 22:1002-11.
32. Horine CF, Warner CG. Experimental occlusion of the pulmonary artery. An anatomic study. Arch Surg 1934; 28: 139-49.
33. Van Allen CM, Nocoll GL, Tuttle WM. Lung changes after occlusion of pulmonary artery branches by embolus and by ligature. Yale J Biol Med 1930; 2: 363-89.
34.Charan NB, Carvalho P. Angiogenesis in bronchial circulatory system after unilateral pulmonary artery obstruction. J Appl Physiol 1997; 82:284-91.
35.Heath D, Edwards JE Jr. The pathology of hypertensive pulmonary vascular disease: a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 1958; 18:533-47.
36.Marshall BE, Marshall C. Pulmonary Hypertension. In: Crystal RG, et al, eds. The Lung. New York: Raven Press Ltd; 1991. p.1177-87.
37.Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD. Pulmonary hemodynamics modify the rat pulmonary artery response to injury. A neointimal model of pulmonary hypertension. Am J Pathol 1997; 151:1019-25.
38.Davies PF, Tripathi SC. Mechanical stress mechanisms and the cell. An endothelial paradigm. Circ Res 1993; 72:239-45.
39.Voelkel NF, Tuder RM. Hypoxia-induced pulmonary vascular remodeling: a model for what human disease? J Clin Invest 2000; 106:733-8.
40.Kim H, Yung GL, Marsh JJ, Konopka RG, Pedersen CA, Chiles PG, et al. Endothelin mediates pulmonary vascular remodeling in a canine model of chronic embolic pulmonary hypertension. Eur Respir J 2000; 15:640-8.
41.Shure D. Thromboendarterectomy: some unanswered questions. Ann Thorac Surg 1996; 62:1253-4.
42.Langille BL, O'Donnell F. Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 1986; 231:405-7.
43.Endrys J, Hayat N, Cherian G. Comparison of bronchopulmonary collaterals and collateral blood flow in patients with chronic thromboembolic and primary pulmonary hypertension. Heart 1997; 78: 171-6.
44.Kauczor HU, Schwickert HC, Mayer E, Schweden F, Schild HH, Thelen M. Spiral CT of bronchial arteries in chronic thromboembolism. J Comput Assist Tomogr 1994; 18:855-61.
45.Haworth SG, de Leval M, Macartney FJ. Hypoperfusion and hyperperfusion in the immature lung. Pulmonary arterial development following ligation of the left pulmonary artery in the newborn pig. J Thorac Cardiovasc Surg 1981; 82:281-92.














 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket