ABSTRACT
Idiopathic pulmonary fibrosis is a type of chronic fibrosing interstitial pneumonia, of unknown etiology, which is associated with a progressive decrease in pulmonary function and with high mortality rates. Interest in and knowledge of this disorder have grown substantially in recent years. In this review article, we broadly discuss distinct aspects related to the diagnosis and treatment of idiopathic pulmonary fibrosis. We list the current diagnostic criteria and describe the therapeutic approaches currently available, symptomatic treatments, the action of new drugs that are effective in slowing the decline in pulmonary function, and indications for lung transplantation.
Keywords:
Idiopathic pulmonary fibrosis/diagnosis; Idiopathic pulmonary fibrosis/therapy; Idiopathic pulmonary fibrosis/rehabilitation.
RESUMO
A fibrose pulmonar idiopática é um tipo de pneumonia intersticial crônica fibrosante, de causa desconhecida, associada à piora funcional respiratória progressiva e taxas elevadas de mortalidade. Em anos recentes, o interesse e os conhecimentos sobre essa moléstia têm aumentado substancialmente. O presente artigo de revisão aborda de maneira ampla aspectos relacionados ao diagnóstico e tratamento da doença. Nele são listados os critérios atuais para o diagnóstico e são discutidos as diferentes abordagens terapêuticas agora disponíveis, o tratamento sintomático, a ação de novas drogas eficazes em reduzir o ritmo de deterioração funcional pulmonar e as indicações para transplante pulmonar.
Palavras-chave:
Fibrose pulmonar idiopática/diagnóstico; Fibrose pulmonar idiopática/terapia; Fibrose pulmonar idiopática/reabilitação.
CONCEITOA fibrose pulmonar idiopática (FPI) é uma forma específica de pneumonia intersticial idiopática crônica, fibrosante e de caráter progressivo. Ela ocorre primariamente em adultos idosos, predominantemente nas sexta e sétima décadas, além de ser restrita aos pulmões. O padrão histológico e/ou radiológico associado à FPI é o de pneumonia intersticial usual (PIU).(1-3) Uma vez confirmado o padrão histológico de PIU associado à FPI, se estabelece um prognóstico significativa-mente pior do que o observado em outras pneumonias intersticiais crônicas.(4) Daí a necessidade do estabelecimento de diagnósticos acurados de FPI, o que, sem dúvida, é um processo desafiador.
Pacientes com FPI exibem mediana de sobrevida de 50% em 2,9 anos, a partir do momento do diagnóstico. (1,4) Contu-do, diante das possibilidades variadas que a história natural da doença pode mostrar, é difícil firmar previsões prognósti-cas acuradas para um paciente com moléstia recém-diagnosticada.(5)
ASPECTOS DIAGNÓSTICOSPara o diagnóstico definitivo de FPI é necessário uma abordagem multidisciplinar integrada, envolvendo pneumologis-tas, radiologistas e patologistas. O diagnóstico de FPI é baseado na ausência de uma causa conhecida de fibrose pulmo-nar, associada à presença de padrão PIU. Esse é o aspecto chave no processo de diagnóstico. Mesmo quando a biópsia pulmonar cirúrgica (BPC) revela o padrão histológico de PIU, é necessário excluir outras condições clínicas que se asso-ciam com esse padrão, incluindo doenças do tecido conectivo, pneumonite de hipersensibilidade na fase crônica (PHC), lesão pulmonar por drogas, asbestose, fibrose pulmonar familiar e síndrome de Hermansky-Pudlak.(1,6,7)
Em 2011, as diretrizes publicadas conjuntamente por American Thoracic Society (ATS), European Respiratory Society (ERS), Japanese Respiratory Society (JRS) e Asociación Latinoamericana del Tórax (ALAT) recomendam, para o diagnós-tico da FPI, uma combinação de critérios envolvendo aspectos de TCAR com características histopatológicas.(1) Esse documento fortalece conclusões prévias de que a TCAR tem um papel primário no diagnóstico de FPI. Em um contexto clínico apropriado, um padrão de PIU definitivo à TCAR elimina a necessidade de BPC (Quadro 1). Entretanto, em pacien-tes apresentando TCAR com padrão possível ou inconsistente para PIU, recomenda-se a BPC (Quadro 2). As combinações específicas de padrões histopatológicos com os observados à TCAR também podem fornecer o grau de probabilidade do diagnóstico de FPI como definitivo, provável ou possível (Quadro 3).(1)

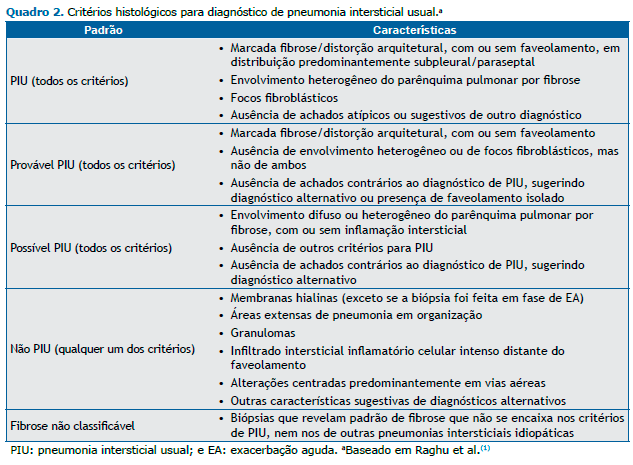

Após a divulgação das diretrizes internacionais,(1) algumas questões surgiram em publicações sequenciais. Inicialmen-te, nas diretrizes brasileiras de doenças pulmonares intersticiais,(6) é discutido se o padrão de PIU possível na TCAR, combinado com a BPC demonstrando PIU possível ou fibrose não classificável, possa ser realmente considerado como FPI provável. A questão levantada foi se a presença de fibrose homogênea com inflamação, como descrito na PIU possí-vel, é realmente compatível com o padrão histológico PIU.(6) Além disso, casos de fibrose não classificável podem ser secundários a erros de amostragem ou a outras condições clínicas que não FPI. Outro ponto relevante é a possibilidade de uma TCAR inconsistente com PIU ainda poder ser compatível com FPI. A presença de opacidades em vidro fosco em regiões basais e subpleurais, observadas principalmente na forma acelerada da doença, se associadas à BPC com PIU definitiva ou provável pode, dentro do contexto apropriado, ser compatível com FPI. Esse conjunto de fatos fez com que as recomendações diagnósticas daquele documento(6) diferissem em algum grau das contidas nas diretrizes internacio-nais.(1) A abordagem brasileira não emprega a classificação de FPI em definitiva, provável ou possível (Quadro 4). Embo-ra mais práticos, esses critérios ainda não ganharam plena aceitação em nosso meio.

Posteriormente, foi ainda evidenciado, em uma casuística espanhola, que 20 de 46 pacientes inicialmente diagnostica-dos com FPI, de acordo com as diretrizes ATS/ERS/JRS/ALAT de 2011,(1) quando revistos em um centro especializado, acabaram por preencher critérios para PHC.(8) Embora esses resultados levantem preocupações quanto ao grau de confi-abilidade fornecido por diagnósticos de PIU baseados unicamente em TCAR, eles não foram considerados suficientemen-te convincentes para alterar as recomendações diagnósticas publicadas naquelas diretrizes.(1) De qualquer modo, tais resultados ressaltam a importância da rigorosa pesquisa de exposições ambientais em todo paciente com doença intersti-cial pulmonar.(9)
DIAGNÓSTICO DIFERENCIALEm pacientes com suspeita de FPI, uma análise cuidadosa deve ser feita quanto à possibilidade de diagnósticos alterna-tivos. Pacientes com padrões tomográficos de PIU provável ou possível são frequentemente encontrados na prática clíni-ca, e o diagnóstico diferencial deve ser feito principalmente contra PHC e pneumonia intersticial não específica fibrótica. Entretanto, uma proporção desses pacientes não irá se submeter à BPC, seja pela presença de contraindicações (comor-bidades, idade avançada ou gravidade da doença), seja por recusa dos indivíduos ao procedimento cirúrgico.
Nesse contexto, a realização de broncoscopia com coleta de lavado broncoalveolar pode ser útil para aumentar o índice de suspeita para o diagnóstico alternativo de PHC, na qual se pode observar linfocitose geralmente acima de 30%.(1,6) Vale ressaltar que biópsias transbrônquicas não são úteis quando a suspeita diagnóstica é de PIU. Todavia, dados recen-tes sugerem que a técnica emergente de criobiópsia endoscópica possa vir a se mostrar útil nesse cenário.(10)
Deve-se lembrar ainda da importância de se excluir o acometimento pulmonar por doenças colágeno-vasculares em pacientes com doenças intersticiais fibrosantes, como a artrite reumatoide e a esclerose sistêmica, mesmo nos pacientes cuja TCAR seja compatível com PIU, principalmente na presença de queixas sugestivas ou história familiar de doenças autoimunes.(7) Vale lembrar ainda a importância da pesquisa de quadros pulmonares semelhantes em parentes, mesmo sendo distantes, devido à ocorrência não rara de pneumopatias intersticiais de incidência familiar.
O diagnóstico precoce e acurado de FPI é um desafio. A ausculta de estertores finos holo e teleinspiratórios, particular-mente do tipo "velcro", é um importante sinal de alerta para essa possibilidade.(11) Atualmente, parece ser mais frequente o excesso de diagnósticos feitos por TCAR, sendo necessário um trabalho urgente de conscientização para a padroniza-ção das abordagens diagnósticas pelos profissionais especializados. Dificuldades ainda existem na avaliação tomográfica como, por exemplo, a distinção entre faveolamento e bronquiectasias de tração, ou entre faveolamento e a combinação de fibrose e enfisema.(12) A integração dos dados de TCAR e histologia é quase sempre útil, mas não atende às necessi-dades de todos os pacientes. A realização de reuniões multidisciplinares é sempre fundamental para o estabelecimento de diagnósticos acurados em pacientes nos quais a aplicação dos critérios propostos se mostrar difícil.
MUDANÇAS DE PARADIGMAS E INSUCESSOS TERAPÊUTICOSO tratamento da FPI foi inicialmente direcionado ao modelo binominal conhecido de inflamação e fibrose; ou seja, uma lesão ou dano desencadearia a inflamação e o processo de reparo pulmonar seria feito com o estabelecimento de fibro-se.(1) Contudo, na FPI, a inflamação identificada é de pequena monta ou praticamente inexistente, e a fibrose é exuberan-te e progressiva. Foi a partir da mudança de paradigma acerca da patogenia da doença, agora considerada como um distúrbio primariamente epitélio-mesenquimal fibrosante, que investigações com novas modalidades terapêuticas foram desenvolvidas.(13)
Além disso, até o ano de 2000, o termo FPI apresentava variações de denominações (alveolite fibrosante, pneumonia crônica idiopática e pneumonite fibrosante) e englobava diversos padrões histológicos. Naquele ano, houve a publicação de um consenso internacional equalizando as variações e definindo com precisão a doença.(14) Assim, todos os estudos publicados anteriormente a 2000 exibem sérias limitações por não terem empregado os critérios atuais de definição da moléstia. Outro aspecto, de muita relevância, foi o desenvolvimento de estudos randomizados com cálculos amostrais efetivos e de preferência multicêntricos, em contrapartida às antigas séries de casos limitadas e trabalhos restritos a um único centro.
A nova fase de investigações do tratamento da FPI cursou com inúmeros ensaios clínicos com resultados negativos, re-sumidos no Quadro 5. O aprendizado adquirido sobre algumas medicações merece atenção especial.
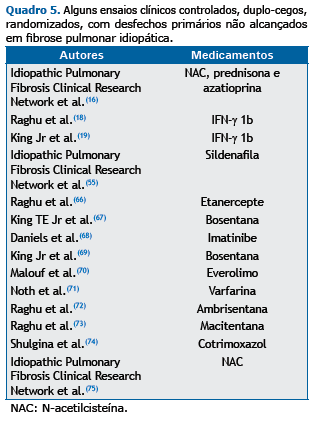
Ao longo de décadas, os corticosteroides acabaram se tornando a terapia padrão para as "fibroses pulmonares", apoia-dos em trabalhos retrospectivos, com poucos pacientes e sem a correta definição de qual condição pulmonar estava sendo tratada.(15) Embora os corticosteroides mostrem-se bastante úteis em alguns quadros intersticiais pulmonares, tais como pneumonia intersticial não específica e pneumonia em organização criptogênica, não há evidências que favoreçam seu uso para o tratamento específico da FPI. Além dessa classe de medicação cursar com muitos efeitos colaterais e elevar o número de comorbidades, há sugestões de que seu uso possa estar associado a uma maior ocorrência de exa-cerbações agudas (EAs).(15)
Os imunossupressores azatioprina e ciclofosfamida, frequentemente utilizados em pneumopatias intersticiais associa-das a doenças colágeno-vasculares, também não estão indicados para pacientes com FPI. Na verdade, em um braço de um estudo randomizado, duplo-cego e placebo-controlado, a combinação de corticosteroides em baixas doses, N-acetilcisteína (NAC) e azatioprina cursou com taxas de hospitalização e de mortalidade significantemente superiores às do uso de placebo. (16) Deve-se admitir que a imunossupressão causada pela combinação das baixas doses de prednisona e azatioprina deva ter influenciado substancialmente tais resultados. Portanto, o emprego de imunossupressores em FPI está atualmente proscrito.
O IFN- é uma citocina endógena que tem propriedades antifibrosantes, imunomoduladoras e antiproliferativas. Inú-meros estudos em modelos animais apontaram para o potencial papel terapêutico do agente em doenças fibrosantes pulmonares. Além disso, um estudo preliminar publicado em 1999 também sugeriu algum grau de eficácia clínica.(17) Esses resultados desencadearam a realização de dois estudos multicêntricos, randomizados, controlados e duplo-cegos, cujos resultados foram desapontadores. (18,19) A importância desses estudos foi mostrar que grandes ensaios clínicos placebo-controlados também eram viáveis em pacientes com FPI, assinalando o início de uma nova era nas pesquisas clínicas dirigidas à doença.
A NAC é precursora da glutationa e importante antioxidante endógeno pulmonar. Considera-se que o estresse oxidativo das células epiteliais alveolares seja uma das vias envolvidas na patogenia da FPI, e os níveis de glutationa parecem estar reduzidos em pacientes com FPI. A reposição com NAC aumenta os valores de glutationa sanguínea desses pacien-tes. Um grupo internacional - Idiopathic Pulmonary Fibrosis International Group Exploring N-Acetylcysteine I Annual (IFIGENIA) - sugeriu que a adição de 1.800 mg de NAC a um esquema terapêutico contendo prednisona em baixas doses e azatioprina poderia levar a redução do ritmo de queda da função pulmonar em pacientes com FPI.(20) Limitações metodológicas apontadas naquele estudo(20) levaram ao desenvolvimento de um ensaio clínico randomizado - denomi-nado PANTHER-IPF - da Idiopathic Pulmonary Fibrosis Clinical Research Network, patrocinado pelo National Heart, Lung, and Blood Institute dos EUA, que avaliou o efeito isolado da NAC em comparação ao uso de placebo na FPI.(21) Verificou-se que, após 60 semanas de uso, não houve diferença estatisticamente significativa entre o grupo NAC (queda da CVF de 180 ml) e o grupo placebo (queda da CVF de 190 ml).(21) Além disso, a mortalidade foi semelhante nos dois grupos, assim como as taxas de EAs. Portanto, no momento, não há evidências que suportem o emprego de NAC em altas doses para o tratamento rotineiro de pacientes com FPI.
Os inúmeros ensaios clínicos com resultados negativos, embora frustrantes, foram importantes por proporcionarem o aprendizado acerca da história natural da FPI e a melhor caracterização dos desfechos a serem utilizados em estudos futuros.
TRATAMENTO ESPECÍFICO DA DOENÇA PULMONARApesar de diversas drogas terem sido investigadas em ensaios clínicos randomizados como agentes potenciais para o tratamento da FPI, até o momento, apenas duas substâncias, de fato, mostraram eficácia no tratamento da moléstia: a pirfenidona e o nintedanibe.
As propriedades antifibróticas da pirfenidona vêm sendo investigadas há décadas em diferentes modelos animais.(22) Acumulam-se evidências de que a droga inibe a deposição de colágeno e cursa com proteção da função pulmonar em roedores tratados com bleomicina por via intratraqueal.
Os mecanismos pelos quais a pirfenidona atua parecem ser pleomórficos, mas ainda não estão completamente esclare-cidos. Dados experimentais indicam que a droga diminui a expressão genética de pró-colágenos, TGF-β e PDGF, além de inibir a produção de TNF-α. A droga parece ainda possuir propriedades antioxidantes.(23)
Os primeiros estudos clínicos realizados com pirfenidona envolveram casuísticas pequenas, foram não controlados ou optaram por desfechos de significância clínica duvidosa.(24-26)
Três foram os melhores estudos de desenho controlado, duplo-cegos e randomizados efetuados com pirfenidona. (27-29) No estudo multicêntrico de Taniguchi et al.,(27) 267 pacientes com FPI foram randomizados para receber, ao longo de 52 semanas, placebo ou pirfenidona, nas doses de 1.200 mg/dia ou 1.800 mg/dia. A medicação, em ambas as doses, levou à redução significativa do grau da queda dos valores de CVF, em comparação ao placebo, ao final do estudo (placebo: −0,16 l; dose baixa: −0,08 l; e dose alta: −0,09 l). Além disso, o uso de pirfenidona em altas doses também esteve associado a um aumento significativo do tempo livre de progressão da doença (tempo para queda da função pulmonar ou óbito) em comparação ao grupo placebo.
Sob um programa denominado Clinical Studies Assessing Pirfenidone in IPF: Research of Efficacy and Safety Outcomes (CAPACITY), foram realizados, simultaneamente, dois ensaios clínicos randomizados, duplo-cegos, placebo-controlados.(28) No estudo 004, os pacientes foram randomizados para receber, ao longo de 72 semanas, placebo ou pirfenidona nas doses de 2.403 mg/dia ou de 1.197 mg/dia. O uso de pirfenidona em altas doses levou à redução signifi-cante da intensidade da queda da CVF observada ao final do estudo em comparação ao grupo placebo (−8,0% vs. −12,4%). O efeito terapêutico da droga foi detectado a partir da vigésima quarta semana de tratamento. O uso da medi-cação em doses elevadas levou ainda a maior duração do tempo livre de progressão da doença. No estudo 006, foram comparados pacientes medicados com pirfenidona em doses de 2.403 mg/dia com aqueles tratados com placebo. Não foi observada diferença significativa no ritmo de queda da CVF ao final do estudo, apesar de o efeito terapêutico sobre essa variável ter sido observado entre a décima segunda e a quadragésima oitava semana de tratamento. Quando os dados dos dois estudos foram combinados, o uso de pirfenidona em doses maiores mostrou um efeito terapêutico no ritmo de queda da CVF a partir da décima segunda semana. Ao final do estudo, o grau de queda da CVF no grupo tratado foi signi-ficantemente inferior ao no grupo placebo (−8,5% vs. −11,5%). Do mesmo modo, o tempo livre de progressão da doença foi maior com o uso da medicação.
Os dados conflitantes dos dois estudos CAPACITY no tocante ao ritmo de queda da CVF, seu desfecho primário, levaram à realização de um ensaio clínico denominado Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND).(29) Naquele estudo, 277 pacientes com FPI receberam placebo, enquanto 278 foram medi-cados com pirfenidona na dose de 2.403 mg/dia por 52 semanas. O uso de pirfenidona esteve associado a um menor ritmo de queda da CVF e a um maior tempo livre de progressão da doença. Ao final do estudo, o grupo controle mostrou uma média de queda na CVF de 428 ml, enquanto, no grupo medicado, esse valor foi de 235 ml. Quando os dados de mortalidade dos estudos CAPACITY foram analisados em conjunto com os do ASCEND, as taxas de mortalidade, por qualquer causa e por FPI, nos grupos pirfenidona foram significativamente inferiores às nos grupos placebo.(28,29)
As cápsulas de pirfenidona contêm 267 mg do sal, o que corresponde a 200 mg da substância ativa. De acordo com o fabricante, a pirfenidona deve ser tomada nas doses de um comprimido de 267 mg v.o. a cada 8 h por uma semana. Na segunda semana, a dose aumenta para duas cápsulas v.o. a cada 8 h e, a partir do décimo quinto dia, passa para três cápsulas v.o. a cada 8 h. A medicação deve ser tomada preferencialmente com alimentos para diminuir o risco de náu-seas. Adequações podem ser feitas em função do surgimento de efeitos adversos, dentre os quais os mais comumente descritos foram náuseas, sintomas dispépticos, erupções cutâneas, fotossensibilidade e alterações de enzimas hepáti-cas.(28,29)
O nintedanibe foi inicialmente chamado de BIBF 1120. A molécula é um derivado da família das indolinonas, que foi desenvolvida originalmente como um agente inibidor da angiogênese para ser empregado no campo da oncologia.(30) A droga já foi testada para o tratamento de tumores sólidos de diferentes linhagens, e sua eficácia clínica já havia sido demonstrada, especialmente para câncer de pulmão não pequenas células.(31)
Os mecanismos pelos quais o nintedanibe age na FPI envolvem a inibição da atividade de receptores cuja ação depen-de de tirosina quinases.(32) A droga bloqueia pontos intracelulares de ligação do ATP em tirosina quinases específicas. Como consequência, ocorre a inativação de receptores celulares para mediadores envolvidos no desenvolvimento da fibrose pulmonar, em especial os receptores para FGF e PDGF. Além disso, o nintedanibe também inibe a ação de recep-tores para VEGF. Como consequência, ocorre prejuízo na proliferação de fibroblastos e redução da deposição de matriz extracelular.
O primeiro ensaio clínico randomizado publicado sobre o uso de nintedanibe em FPI denominou-se To imprOve pulMO-naRy fibROsis With BIBF1120 (TOMORROW) e teve uma duração de 12 meses. Naquele estudo,(33) 432 pacientes foram randomizados para receber placebo ou quantidades crescentes da medicação, culminando em 150 mg duas vezes ao dia. O uso de nintedanibe nas doses de 150 mg, duas vezes ao dia, cursou com redução significativa do número de episódios de EAs em comparação ao de placebo. Além disso, ao final do estudo, a intensidade de queda da CVF no grupo tratado com essas doses da droga foi menor do que a do grupo controle (−0,06 l vs.−0,19 l; p = 0,06).
Dois ensaios de fase III adicionais, relacionados à eficácia do nintedanibe na FPI, foram desenvolvidos simultaneamen-te e denominados INPULSIS.(34) Em ambos os ensaios, a dose empregada da medicação foi de 150 mg duas vezes ao dia. No ensaio INPULSIS-1, o uso de nintedanibe levou a uma redução significativa da taxa anual de queda da CVF em com-paração ao de placebo (−114,7 ml vs. −239,9 ml). No estudo INPULSIS-2, o uso da medicação também cursou com redu-ção significativa da taxa anual de queda da CVF em relação ao de placebo (−113,6 ml vs. −207,3 ml). Naquele estudo, o uso de nintedanibe também esteve associado a um aumento significativo do tempo para surgimento do primeiro episódio de EA.
A dose de nintedanibe recomendada pelo fabricante é de 150 mg, v.o., duas vezes ao dia. Essa dose pode ser reduzida transitoriamente para 100 mg/dia em função do surgimento de reações adversas. Recomenda-se ingerir a droga com um copo cheio d'água e comida. Os efeitos adversos mais comumente associados ao uso da medicação são de natureza digestiva, em especial, diarreia e náuseas. A diarreia está presente em aproximadamente 62% dos pacientes em uso de nintedanibe, mas costuma ser controlada com a associação de loperamida.
Ainda que a pirfenidona já tivesse sido aprovada para venda no Japão e na Europa em função de estudos prévios, a agência americana Food and Drug Administration apenas a liberou para uso nos EUA após os resultados do ensaio AS-CEND. Na mesma data, o nintedanibe também foi aprovado para uso no país. A aprovação das duas drogas pela Food and Drug Administration baseou-se, fundamentalmente, no efeito benéfico observado sobre o ritmo de queda da CVF.(35) Mesmo que a CVF seja um fator de prognóstico na FPI, o ideal seria identificar um efeito positivo das novas drogas dire-tamente sobre a mortalidade dos pacientes. Infelizmente, a caracterização desses efeitos deverá demandar o acompa-nhamento de um maior número de pacientes por períodos prolongados.
Análises de seguimento de longo prazo ligadas aos ensaios clínicos citados foram recentemente divulgadas. Há agora sugestões de que a manutenção de pirfenidona, mesmo em pacientes que exibiram queda de pelo menos 10% da CVF ao final de 6 meses de tratamento, esteve associada a uma melhor evolução do que naqueles em uso de placebo. Além disso, o uso de nintedanibe por até 76 semanas mostrou o mesmo perfil de eficácia e de efeitos adversos do observado no seu uso por 52 semanas. Também há sugestões de que ambas as drogas sejam eficazes em pacientes com FPI menos avançada.
Devido a esse conjunto de dados, a atualização de 2015 da diretriz das sociedades ATS/ERS/JRS/ALAT sobre o trata-mento de FPI sugere o uso de pirfenidona ou de nintedanibe como opções terapêuticas para o tratamento da moléstia.(36)
É importante ressaltar que, no presente momento, as evidências quanto à eficácia de ambas as drogas se restringem à FPI, mas não a outras formas de doenças intersticiais pulmonares fibrosantes, tais como PHC ou comprometimento pul-monar por doenças colágeno-vasculares. Além disso, no presente momento, não há indicação para o uso das duas dro-gas em associação, ainda que um estudo inicial tenha sugerido a segurança dessa.(37) Nesse contexto, a opção por uma ou outra medicação deve ser feita caso a caso, e basear-se em aspectos como disponibilidade do produto no mercado, comorbidades, aderência e tolerabilidade dos pacientes aos efeitos adversos, ou ainda a falha prévia do uso de terapia.
Uma questão adicional, ainda não esclarecida, relaciona-se ao melhor momento de introdução da medicação. A maioria dos especialistas no assunto sugere começar o tratamento da FPI com alguma das duas novas drogas assim que o diag-nóstico for estabelecido. A justificativa baseia-se no mau prognóstico habitualmente associado com a doença e a possibi-lidade do surgimento de EAs a qualquer momento. Sendo assim, o uso da medicação estaria justificado mesmo naqueles poucos pacientes diagnosticados com função pulmonar normal. Entretanto, alguns especialistas não compartilham dessa última opinião, alegando que é difícil se estabelecer o prognóstico individual para um determinado paciente, além de as EAs serem mais comuns na fase avançada da doença. Nesse contexto, poder-se-ia monitorar o comportamento da função pulmonar por algum tempo, e a introdução da medicação dar-se-ia assim que detectada alguma deterioração. Essa é uma questão polêmica, no momento sem resposta definitiva, ficando a critério do clínico e do paciente a tomada conjunta de decisão. Finalmente, devido aos critérios de exclusão utilizados pelos estudos, a real efetividade das novas medicações para pacientes com doença muito avançada também não está determinada.
TRATAMENTO SINTOMÁTICOTosseA tosse é um sintoma bastante frequente e seu controle pode se tornar difícil nos pacientes com FPI, contribuindo de modo significativo para a piora da qualidade de vida. Ela pode estar relacionada ao refluxo gastroesofágico (RGE); po-rém, na maioria das vezes, é secundária à própria FPI, sendo mais comum nos indivíduos com doença mais avançada.(6)
O tratamento empírico do RGE pode levar à melhora da tosse em alguns casos. Poucas opções estão disponíveis para o tratamento da tosse relacionada à doença propriamente dita, quando antitussígenos tradicionais, como a codeína, falham. Apesar da ausência de efeito sobre a evolução da doença, corticosteroides (prednisona, 20-30 mg/dia) podem promover alívio desse sintoma.(1,6) Um estudo randomizado demonstrou que a talidomida (50-100 mg/dia v.o.), um derivado do ácido glutâmico, determina uma melhora da tosse e da qualidade de vida em pacientes com FPI. Contudo, a referida droga ainda não está legalmente aprovada para esse uso no nosso meio.(38) Há sugestões que a gabapentina (300-1.800 mg/dia) também poderá se mostrar útil para o tratamento dessa condição.(39)
DispneiaA dispneia progressiva é bastante comum nos pacientes com FPI, relacionando-se com pior qualidade de vida e maior risco de depressão e de óbito; frequentemente é de difícil controle.(40) A origem da dispneia na FPI envolve a própria evolução da doença, além da contribuição de outros fatores, como depressão, ansiedade e fraqueza muscular. Apesar das evidências limitadas, morfina v.o., geralmente em baixas doses (até 20 mg/dia), pode ser utilizada em casos selecio-nados, ajustando-se a ingestão de acordo com a resposta e com a presença de efeitos adversos, como sonolência e cons-tipação intestinal. (6,41) Para pacientes hipoxêmicos, a suplementação de oxigênio ao repouso ou durante o esforço pode determinar alívio da dispneia (Quadro 6). A reabilitação pulmonar também pode contribuir para a redução do grau de dispneia.(42)
 Depressão e ansiedade
Depressão e ansiedadeMuitos pacientes com FPI apresentam sintomas de depressão e ansiedade, que devem ser rotineiramente pesquisados nessa população. A prevalência de depressão varia entre 25% e 50% dos pacientes com FPI, relacionando-se com maior grau de dispneia e maior limitação funcional.(42) A ansiedade ocorre em torno de 30-40% dos casos, associando-se tam-bém com maior intensidade de dispneia.(42) Nesse contexto, apesar da ausência de estudos robustos comprobatórios, a abordagem da dispneia pode determinar uma melhora dos sintomas de depressão e ansiedade. Pode-se considerar ainda a realização de acompanhamento psicológico e a utilização de agentes ansiolíticos e antidepressivos.
RECONHECIMENTO DE COMORBIDADESAlgumas comorbidades têm sido identificadas com muita frequência em pacientes com FPI. A explicação para isso pode residir, pelo menos em parte, no fato de que pacientes com FPI são idosos e comumente ex-fumantes. Nesse contexto, a identificação e o tratamento de comorbidades poderão contribuir para a melhora da qualidade de vida e mesmo da so-brevida.
RGEA descrição de uma relação entre a FPI e eventos de microaspiração de conteúdo gástrico não é recente e vem sendo reforçada por dados biológicos e clínicos. (43) Substâncias presentes no conteúdo gástrico, como a pepsina, já foram en-contradas em níveis elevados em amostras de lavado broncoalveolar de pacientes com EA da FPI.(43) A presença de hérnia de hiato em estudos tomográficos do tórax parece ser mais frequente em FPI do que em outras doenças pulmona-res, como asma e DPOC. A presença de refluxo ácido detectado através de monitorização do pH também foi frequente em estudos de duas diferentes coortes. Inicialmente, em 65 pacientes com FPI, foi relatada a presença de refluxo ácido em 87% daqueles pacientes, frequência mais elevada que nos controles com asma utilizados no estudo.(44) Em outra investi-gação, foi relatada a frequência de 94% de refluxo ácido em pacientes com FPI comparada com 50% em pacientes com outras doenças intersticiais que serviram como controle.(45) Contudo, naqueles dois estudos,(44,45) apenas 47% e 25% dos pacientes com FPI, respectivamente, apresentaram sintomas clássicos sugestivos de RGE. Esses dados reforçam que os sintomas de RGE não devem ser usados como ferramenta de rastreamento.
Não existem ensaios clínicos controlados avaliando os efeitos do tratamento do refluxo em pacientes com FPI. Em uma pequena série de 4 pacientes com FPI e RGE tratados com inibidores de bomba de prótons (IBP) ou fundoplicatura gástri-ca, observou-se estabilização da função pulmonar.(46) Resultados semelhantes foram encontrados em uma série de paci-entes com FPI aguardando transplante pulmonar e que realizaram fundoplicatura para o tratamento de RGE sintomáti-co. (47) A análise dos dados dos grupos placebo de três ensaios clínicos em FPI mostrou que o uso das medicações antiáci-das com IBP ou antagonistas de receptores H2 por 30 semanas se associou a menor ritmo de redução da CVF e menor frequência de EAs. (48) Em um estudo retrospectivo com 204 pacientes acompanhados em dois centros médicos nos EUA, tanto o uso de medicações antirrefluxo como a realização de fundoplicatura gástrica cursaram com aumento significante da sobrevida.(49)
Mesmo que ainda sejam necessários ensaios clínicos bem desenhados e controlados para responder adequadamente a essa questão, o uso de IBP e/ou antagonistas de receptores H2 parece ser benéfico para pacientes com FPI, mesmo na ausência de sintomas. O mesmo parece ser verdade para o procedimento de fundoplicatura gástrica em casos seleciona-dos. Devido a isso, a atualização de 2015 das sociedades ATS/ERS/JRS/ALAT reafirma a recomendação já feita em 2011 para o tratamento rotineiro do RGE em pacientes com FPI.(36)
Câncer de pulmãoPacientes com FPI apresentam um risco mais elevado de apresentar carcinoma de pulmão que a população geral. Em um estudo com 890 pacientes com diagnóstico de FPI, foi relatado um risco 7,31 vezes maior nesses pacientes que em indivíduos da população geral, independentemente da história de tabagismo. (50) Os fatores etiopatogênicos dessa associ-ação ainda não estão perfeitamente estabelecidos, e a detecção radiológica pode ser dificultada pelas anormalidades fibróticas previamente existentes. A coexistência dessas entidades interfere no tratamento de ambas, com risco de com-plicações operatórias, exacerbações e toxicidade pulmonar por uso de medicamentos e por radioterapia.
Distúrbios do sonoPacientes com FPI podem apresentar dessaturação durante o sono, independentemente da presença de apneia obstruti-va do sono (AOS). A qualidade do sono nesse grupo de pacientes pode ser prejudicada pela presença de RGE, tosse noturna, uso de medicações, ou mesmo AOS.(51) Pacientes portadores de FPI apresentam ainda pior qualidade do sono, maior frequência de AOS e dessaturação noturna que indivíduos saudáveis da mesma faixa etária.
Em um estudo, o diagnóstico de AOS foi confirmado por polissonografia em 88% dos pacientes com FPI, a maior parte deles com doença moderada ou grave. (52) Investigações adicionais foram feitas sobre o uso de continuous positive air-way pressure (CPAP, pressão positiva contínua nas vias aéreas) noturna em pacientes com diagnóstico recente de FPI e AOS moderada ou grave.(53) Os resultados foram analisados com os sujeitos divididos entre pacientes com boa ou com má aceitação ao tratamento. Os dois grupos exibiram melhoras de qualidade de vida e do sono, com menor intensidade nos indivíduos com má aceitação. Adicionalmente, os pacientes com boa aceitação ao tratamento apresentaram sobrevi-da maior do que os do outro grupo ao longo do período de avaliação. Por tudo isso, a AOS deve ser ativamente pesqui-sada e, quando indicado, o tratamento com CPAP deve ser introduzido.
Hipertensão pulmonarA hipertensão pulmonar é uma complicação bem reconhecida em pacientes com FPI, particularmente nas fases avança-das da doença. Em pacientes aguardando transplante pulmonar, a prevalência é de 46,1%.(54) Na maioria das vezes, o grau da hipertensão é moderada; porém, em aproximadamente 9% dos casos, ela pode ser grave (pressão média da artéria pulmonar ≥ 35 mmHg ou essa pressão ≥ 25 mmHg com índice cardíaco < 2 l/min/m2).
Níveis elevados de hipertensão pulmonar estão associados com pior sobrevida.(1,6) Ensaios clínicos que empregaram medicações para redução da hipertensão pulmonar em pacientes com FPI não atingiram os seus desfechos primá-rios.(55,56) Apesar disso, em um estudo, o uso de sildenafila esteve associado com melhora da PaO2, DLCO, intensidade da dispneia e qualidade de vida.(55) Uma análise post hoc dos dados do último estudo sugere que a droga poderia ser mais efetiva em pacientes com evidências ecocardiográficas de disfunção sistólica do ventrículo direito.(57) Embora no momen-to não haja fortes evidências que apoiem o uso rotineiro de medicações para reduzir a hipertensão pulmonar em pacien-tes com FPI, a questão não parece estar completamente esclarecida.
Enfisema pulmonarA FPI e o enfisema compartilham o tabagismo como fator de risco, e a prevalência de enfisema em pacientes com FPI varia entre 30% e 55%. Embora já descrita em anos anteriores, persiste o debate se essa seria uma entidade clínica específica, com base genética distinta, ou uma coincidência em pacientes tabagistas. (58) A maioria dos indivíduos é do sexo masculino, com preservação dos volumes pulmonares e importante redução da DLCO. Pacientes com a combinação FPI e enfisema exibem um prognóstico pior do que pacientes com FPI isolada. Neles, a frequência de hipertensão pulmo-nar é maior e cursa com maior influência na sobrevida que a redução dos volumes pulmonares.(12,59) A condução tera-pêutica específica para esses pacientes ainda é incerta pela falta de dados na literatura, baseando-se na suplementação de oxigênio, cessação do tabagismo e medidas gerais.
Doenças cardiovascularesO risco de aparecimento de doenças cardiovasculares parece ser maior em pacientes com FPI.(1,6) Um estudo que ava-liou 920 pacientes com FPI demonstrou riscos elevados de angina, trombose venosa profunda e eventos coronarianos agudos antes do diagnóstico. (60) Após o diagnóstico, trombose venosa profunda e doença coronariana aguda apresenta-ram riscos relativos especialmente elevados (3,39 e 3,14, respectivamente). Portanto, a avaliação regular de eventos cardiovasculares e tromboembólicos deve fazer parte da condução de pacientes com FPI, tanto na fase estável como nas EAs.
TRATAMENTOS NÃO MEDICAMENTOSOSEducaçãoÉ fundamental que o paciente e seus familiares sejam continuamente informados sobre diversos aspectos da doença, incluindo fisiopatologia, sintomas, evolução, tratamento, incluindo medidas paliativas, para a melhora de qualidade de vida e do prognóstico. Adicionalmente, quando pertinentes, questões relativas à terminalidade da vida devem ser abor-dadas. Nesse contexto, para o manejo mais adequado, preferências e crenças do paciente devem ser valorizadas e discu-tidas com os profissionais que o acompanham.
VacinaçãoNão há estudos que tenham avaliado o impacto da utilização de vacinas especificamente em pacientes com FPI. Entre-tanto, recomenda-se a aplicação das vacinas contra influenza (anualmente) e antipneumocócica nessa população.(6)
Suplementação de oxigênioA hipoxemia é um evento bastante comum nos pacientes com FPI, e muitos necessitarão de oxigênio suplementar du-rante a evolução da doença. Todos os pacientes deverão ser periodicamente avaliados quanto à ocorrência de hipoxemia ao repouso e durante o esforço.(1,6) Apesar da ausência de estudos randomizados que tenham avaliado o impacto da suplementação de oxigênio na mortalidade em pacientes com FPI, recomenda-se sua utilização nas situações listadas no Quadro 6.
Sugere-se utilizar fluxo de oxigênio para se manter a SpO2 entre 90% e 92%, podendo-se observar inclusive melhora do desempenho aos esforços. Foi demonstrado que a necessidade de suplementação de oxigênio se relaciona ao prog-nóstico, de modo que quanto maior o fluxo de oxigênio necessário ao repouso, menor a sobrevida.(1,6) Recomenda-se avaliar ainda a necessidade de suplementação de oxigênio durante viagens aéreas.
Reabilitação pulmonarA limitação ao exercício de grau variado é frequentemente observada nos pacientes com FPI, apresentando múltiplas causas, que atuam de maneira isolada ou combinada, como alterações nas trocas gasosas, limitações ventilatórias, hiper-tensão pulmonar e disfunções da musculatura periférica. Um programa de reabilitação pulmonar (RP) envolve treinamen-to aeróbico e melhora da força muscular, além de educação sobre a doença, orientação nutricional e suporte psicossocial. Apesar do número limitado de estudos robustos que avaliaram o impacto da RP em pacientes com FPI, a RP pode produ-zir redução do grau de dispneia e melhora da qualidade de vida, assim como aumento do tempo de exercício e da distân-cia percorrida no teste de caminhada de seis minutos nesses pacientes.(61) Um estudo recente demonstrou que os benefí-cios da RP podem ser mantidos a longo prazo, trazendo a perspectiva de se realizar essa intervenção terapêutica por períodos mais prolongados nessa população.(62) Portanto, recomenda-se a realização de RP nos pacientes com FPI, com duração de pelo menos 12 semanas, exceto quando houver alguma contraindicação. Trata-se de um procedimento segu-ro e com baixo risco de eventos adversos nesse grupo de pacientes.
Na indisponibilidade de acesso a um programa formal de RP, os pacientes podem ser estimulados a realizarem cami-nhadas com duração entre 20 e 30 minutos, por pelo menos três vezes por semana, com suplementação de oxigênio quando necessário.
Transplante pulmonarO transplante pulmonar é uma alternativa de tratamento que proporciona um aumento da sobrevida aos pacientes com FPI.(1,6) Em função da progressão rápida que ocorre em grande parte dos pacientes, pela idade avançada e pelas comor-bidades associadas, a FPI é a doença que determina a maior taxa de mortalidade entre os pacientes em lista de espera para transplante pulmonar. Deve-se, portanto, estar atento ao momento correto de encaminhamento desses pacientes para avaliação por um centro de transplantes, uma vez que frequentemente ela acaba ocorrendo tardiamente. O ideal é que os pacientes sejam encaminhados para uma avaliação inicial no momento do diagnóstico de FPI, independentemente do grau de disfunção. As indicações e contraindicações do transplante pulmonar para pacientes com FPI estão listadas no Quadro 7.(6,63)

A sobrevida dos pacientes com FPI submetidos a transplante de pulmão fica em torno de 50% em 5 anos, com mediana de 4,5 anos, sendo pior que a observada em indivíduos submetidos a transplante por outras doenças pulmonares.
Apesar de o transplante de pulmão ser uma opção que melhora o prognóstico dos pacientes com FPI, o número de pro-cedimentos realizados em nosso meio é limitado e não atende toda a demanda necessária, uma vez que existem poucos centros de referência capazes de realizá-los, associado ao fato de o número de doadores ainda ser bastante restrito.
EXACERBAÇÕES AGUDASEpisódios de EA da FPI correspondem a um agravamento agudo (inferior a 30 dias) das condições clínicas do paciente, representado por aumento da dispneia e maior necessidade de suplementação de oxigênio, associado ao surgimento de novas imagens pulmonares na TCAR (imagens em vidro fosco ou consolidações pulmonares bilaterais sob um padrão tomográfico anterior de PIU).(1,6,64) Condições clínicas como insuficiência cardíaca, tromboembolismo pulmonar, pneumo-tórax e infecção devem ser afastadas. O quadro deve corresponder a uma exacerbação idiopática da doença, sem causa conhecida. Essa agudização idiopática está associada com um aumento da mortalidade, já tendo sido descrita em até 85% dos casos.(1,6,64)
A incidência da EA é pouco conhecida, em decorrência da falta de uma definição consensual e pelo fato de que, em ge-ral, os estudos que avaliam essa condição são retrospectivos. O tempo de diagnóstico da FPI pode estar associado a um maior risco de EA. Já foi relatado que a proporção de pacientes que apresentam EAs no primeiro ano de doença após o diagnóstico é inferior àquela observada no terceiro ano de doença.
Os fatores de risco identificados como associados à EA incluem CVF < 72% do previsto, DLCO < 62%, nunca ter sido fumante e presença de hipertensão arterial pulmonar.(60) Doenças mais extensas na TCAR possuem um maior risco de mortalidade por EA em 3 meses em comparação a doenças mais limitadas. Uma revisão sistemática demonstrou que o risco de óbito por EA em 1 mês é de 60% e, em 3 meses, é de 67%. Após um quadro de EA, pacientes com FPI sobrevi-vem, em média, 2,2 meses.
A etiologia das EAs é desconhecida. É possível que sejam desencadeadas por uma súbita aceleração da doença fibróti-ca subjacente, infecção de etiologia não reconhecida, infecção viral, procedimentos cirúrgicos torácicos e extratorácicos, realização de broncoscopia ou presença de microaspirações de conteúdo gastroesofágico.
O achado mais comum na TCAR é o aparecimento de novas imagens de opacidades do tipo vidro fosco sobrepostas a infiltrado intersticial reticular e faveolamento pré-existentes. O padrão de distribuição das opacidades parece estar asso-ciado com o prognóstico, cursando com melhor evolução quando pequenas e mais periféricas e com pior desfecho quan-do difusas e multifocais. Na histologia encontra-se dano alveolar difuso sobreposto ao padrão PIU. Tem sido descrito também um padrão de organização pneumônica criptogênica e lesão aguda pulmonar inespecífica sem membrana hiali-na.
O tratamento das EAs ainda está envolto em muitas incertezas. Não existem estudos duplo-cegos, controlados e rando-mizados sobre o tema. Diretrizes de sociedades especializadas recomendam o emprego de corticosteroides e cuidados clínicos intensivos. Essa recomendação, fraca, foi baseada em artigos com baixa qualidade de evidência e não especifica doses, duração do tratamento e via de administração das medicações.(1,6,64)
Outras propostas terapêuticas têm sido estudadas, como a hemoperfusão direta com polimixina B em coluna fibra imo-bilizada, rituximabe ou trombomodulina, além do controle do RGE.(65)
Apesar da falta de claras evidências clínicas, o uso dos corticosteroides se apoia no tipo de lesão pulmonar que aconte-ce na EA. A opção mais frequente são pulsos de metilprednisolona, na dose de 1,0 g por 3 dias consecutivos. Entretanto, a experiência clínica sugere que, em algumas situações, doses menores podem se mostrar igualmente efetivas. Na im-possibilidade da realização de broncoscopia ou de se descartar completamente a presença de infecção, a maioria dos clínicos tende a também empregar antibioticoterapia de largo espectro previamente ou simultaneamente ao uso de corti-costeroides.
PERSPECTIVASUm número substancial de progressos vem ocorrendo no campo da FPI nas últimas décadas. Pela primeira vez, há um conjunto de conhecimentos e disponibilidade de moléculas capazes de claramente influenciar de forma positiva a história natural da moléstia (Quadro 8). Além disso, diversas indústrias farmacêuticas dedicam-se atualmente ao desenvolvimen-to de novas opções terapêuticas para uma doença até recentemente considerada órfã. É sabido que vários estudos em fase II e III envolvendo novas moléculas, inclusive agentes imunobiológicos, estão em andamento, o que é um fato muito alvissareiro.

Podemos então supor que, nas próximas décadas, serão observados aumentos significativos na sobrevida de pacientes com FPI, o que fará crescer o número de pacientes em acompanhamento por essa doença nos centros de atendimento médico especializados.
Apesar desses avanços, um número substancial de necessidades e desafios ainda persiste, entre eles: (i) promoção do aumento de conhecimento acerca da doença no meio médico e na sociedade como um todo; (ii) desenvolvimento de métodos diagnósticos para a doença em fase inicial; (iii) melhor entendimento das bases genéticas da moléstia e do seu modo de interação com agentes ambientais; (iv) caracterização dos dados de incidência, prevalência e padrão de distri-buição da doença nas diferentes partes do mundo; (v) agilização, por parte das agências governamentais, dos processos de aprovação para o uso clínico de drogas com eficácia realmente comprovada; e (vi) garantia de acesso democrático e universal a formas de tratamento medicamentoso e não medicamentoso para os indivíduos acometidos.
É de se supor que a luta contra a FPI só será ganha com uma ampla coalizão de esforços, envolvendo cientistas bási-cos, pesquisadores clínicos, médicos, empresas farmacêuticas, associações representativas de pacientes e familiares e, em muitos países, o imprescindível apoio governamental.
REFERÊNCIAS1. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824. http://dx.doi.org/10.1164/rccm.2009-040GL
2. Kawano-Dourado L, Kairalla RA. Usual interstitial pneumonia: a pattern or a disease? A reflection upon the topic. J Bras Pneumol. 2013:39(1):111-2. http://dx.doi.org/10.1590/S1806-37132013000100017
3. Wells AU. The revised ATS/ERS/JRS/ALAT diagnostic criteria for idiopathic pulmonary fibrosis (IPF)--practical implications. Respir Res. 2013;14 Suppl 1:S2.
4. King TE Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001;164(7):1171-81. http://dx.doi.org/10.1164/ajrccm.164.7.2003140
5. Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142(12 Pt 1):963-7. http://dx.doi.org/10.7326/0003-4819-142-12_Part_1-200506210-00005
6. Baldi BG, Pereira CA, Rubin AS, Santana AN, Costa AN, Carvalho CR, et al. Highlights of the Brazilian Thoracic Association guidelines for interstitial lung diseases. J Bras Pneumol. 2012;38(3):282-91. http://dx.doi.org/10.1590/S1806-37132012000300002
7. Fischer A. Interstitial lung disease in suggestive forms of connective tissue disease. J Bras Pneumol. 39(6): 641-43. http://dx.doi.org/10.1590/S1806-37132013000600001
8. Morell F, Villar A, Montero MÁ, Mu-oz X, Colby TV, Pipvath S, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1(9):685-94. http://dx.doi.org/10.1016/S2213-2600(13)70191-7
9. Cardoso J, Carvalho I. The value of family history in the diagnosis of hypersensitivity pneumonitis. J Bras Pneumol. 2014;40(2):183-7. http://dx.doi.org/10.1590/S1806-37132014000200013
10. Pajares V, Puzo C, Castillo D, Lerma E, Montero MA, Ramos-Barbón D, et al. Diagnostic yield of transbronchial cryobiopsy in interstitial lung disease: a randomized trial. Respirology. 2014;19(6):900-6. http://dx.doi.org/10.1111/resp.12322
11. Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J. 2012;40(3):519-21. http://dx.doi.org/10.1183/09031936.00001612
12. Dias OM, Baldi BG, Costa AN, Carvalho CR. Combined pulmonary fibrosis and emphysema: an increasingly recognized condition. J Bras Pneumol. 2014;40(3):304-12. http://dx.doi.org/10.1590/S1806-37132014000300014
13. Selman M, King TE, Pardo A; American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134(2):136-51. http://dx.doi.org/10.7326/0003-4819-134-2-200101160-00015
14. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646-64.
15. Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev. 2003;(3):CD002880. http://dx.doi.org/10.1002/14651858.cd002880
16. Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968-77. http://dx.doi.org/10.1056/NEJMoa1113354
17. Ziesche R, Hofbauer E, Wittmann K, Petkov V, Block LH. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 1999;341(17):1264-9. http://dx.doi.org/10.1056/NEJM199910213411703
18. Raghu G, Brown KK, Bradford WZ, Starko K, Noble PW, Schwartz DA, et al. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350(2):125-33. http://dx.doi.org/10.1056/NEJMoa030511
19. King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374(9685):222-8. http://dx.doi.org/10.1016/S0140-6736(09)60551-1
20. Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353(21):2229-42. http://dx.doi.org/10.1056/NEJMoa042976
21. Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2093-101. http://dx.doi.org/10.1056/NEJMoa1401739
22. Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125(6):779-85.
23. Carter NJ. Pirfenidone in idiopathic pulmonary fibrosis. Drugs. 2011;71(13):1721-32. http://dx.doi.org/10.2165/11207710-000000000-00000
24. Raghu G, Johnson WC, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label Phase II study. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1061-9. http://dx.doi.org/10.1164/ajrccm.159.4.9805017
25. Nagai S, Hamada K, Shigematsu M, Taniyama M, Yamauchi S, Izumi T. Open-label compassionate use one year-treatment with pirfenidone to patients with chronic pulmonary fibrosis. Intern Med. 2002;41(12):1118-23. http://dx.doi.org/10.2169/internalmedicine.41.1118
26. Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171(9):1040-7. http://dx.doi.org/10.1164/rccm.200404-571OC
27. Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821-9. http://dx.doi.org/10.1183/09031936.00005209
28. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377(9779):1760-9. http://dx.doi.org/10.1016/S0140-6736(11)60405-4
29. King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083-92. Erratum in: N Engl J Med. 2014;371(12):1172. http://dx.doi.org/10.1056/NEJMoa1402582
30. Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774-82. http://dx.doi.org/10.1158/0008-5472.CAN-07-6307
31. Rashdan S, Hanna N. Nintedanib for the treatment of non-small-cell lung cancer. Expert Opin Pharmacother. 2014;15(5):729-39. http://dx.doi.org/10.1517/14656566.2014.897695
32. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1434-45. http://dx.doi.org/10.1183/09031936.00174914
33. Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079-87. http://dx.doi.org/10.1056/NEJMoa1103690
34. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-82. Erratum in: N Engl J Med. 2015;373(8):782. http://dx.doi.org/10.1056/NEJMoa1402584
35. Karimi-Shah BA, Chowdhury BA. Forced vital capacity in idiopathic pulmonary fibrosis--FDA review of pirfenidone and nintedanib. N Engl J Med. 2015;372(13):1189-91. http://dx.doi.org/10.1056/NEJMp1500526
36. Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: Treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3-e19. http://dx.doi.org/10.1164/rccm.201506-1063ST
37. Ogura T, Taniguchi H, Azuma A, Inoue Y, Kondoh Y, Hasegawa Y, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1382-92. http://dx.doi.org/10.1183/09031936.00198013
38. Horton MR, Santopietro V, Mathew L, Horton KM, Polito AJ, Liu MC, et al. Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: a randomized trial. Ann Intern Med. 2012;157(6):398-406. http://dx.doi.org/10.7326/0003-4819-157-6-201209180-00003
39. Inoue K, Takano H. Gabapentin for refractory chronic cough. Lancet. 2013;381(9867):623. http://dx.doi.org/10.1016/S0140-6736(13)60338-4
40. Ryerson CJ, Donesky D, Pantilat SZ, Collard HR. Dyspnea in idiopathic pulmonary fibrosis: a systematic review. J Pain Symptom Manage. 2012;43(4):771-82. http://dx.doi.org/10.1016/j.jpainsymman.2011.04.026
41. Allen S, Raut S, Woollard J, Vassallo M. Low dose diamorphine reduces breathlessness without causing a fall in oxygen saturation in elderly patients with end-stage idiopathic pulmonary fibrosis. Palliat Med. 2005;19(2):128-30. http://dx.doi.org/10.1191/0269216305pm998oa
42. Holland AE, Fiore JF Jr, Bell EC, Goh N, Westall G, Symons K, et al. Dyspnoea and comorbidity contribute to anxiety and depression in interstitial lung disease. Respirology. 2014;19(8):1215-21. http://dx.doi.org/10.1111/resp.12360
43. Lee JS. The role of gastroesophageal reflux and microaspiration in idiopathic pulmonary fibrosis. Clin Pulm Med. 2014;21(2):81-85. http://dx.doi.org/10.1097/CPM.0000000000000031
44. Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, Hayes J, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27(1):136-42. http://dx.doi.org/10.1183/09031936.06.00037005
45. Tobin RW, Pope CE 2nd, Pellegrini CA, Emond MJ, Sillery J, Raghu G. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158(6):1804-8. http://dx.doi.org/10.1164/ajrccm.158.6.9804105
46. Raghu G, Yang ST, Spada C, Hayes J; Pellegrini CA. Sole treatment of acid gastroesophageal reflux in idiopathic pulmonary fibrosis: a case series. Chest. 2006;129(3):794-800. http://dx.doi.org/10.1378/chest.129.3.794
47. Linden PA, Gilbert RJ, Yeap BY, Boyle K, Deykin A, Jaklitsch MT, et al. Laparoscopic fundoplication in patients with end-stage lung disease awaiting transplantation. J Thorac Cardiovasc Surg. 2006;131(2):438-46. http://dx.doi.org/10.1016/j.jtcvs.2005.10.014
48. Lee JS, Collard HR, Anstrom KJ, Martinez FJ, Noth I, Roberts RS, et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomized controlled trials. Lancet Respir Med. 2013;1(5):369-76. http://dx.doi.org/10.1016/S2213-2600(13)70105-X
49. Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(12):1390-4. http://dx.doi.org/10.1164/rccm.201101-0138OC
50. Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med. 2000;161(1):5-8. http://dx.doi.org/10.1164/ajrccm.161.1.9906062
51. Troy LK, Corte TJ. Sleep disordered breathing in interstitial lung disease: A review. World J Clin Cases. 2014;2(12):828-34. http://dx.doi.org/10.12998/wjcc.v2.i12.828
52. Lancaster H, Mason WR, Parnell JA, Rice TW, Loyd JE, Milstone AP, et al. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest. 2009;136(3):772-8. http://dx.doi.org/10.1378/chest.08-2776
53. Mermigkis C, Bouloukaki I, Antoniou K, Papadogiannis G, Giannarakis I, Varouchakis G, et al. Obstructive sleep apnea should be treated in patients with idiopathic pulmonary fibrosis. Sleep Breath. 2015;19(1):385-91. http://dx.doi.org/10.1007/s11325-014-1033-6
54. Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30(4):715-21. http://dx.doi.org/10.1183/09031936.00107206
55. Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620-8. http://dx.doi.org/10.1056/NEJMoa1002110
56. Corte TJ, Keir GJ, Dimopoulos K, Howard L, Corris PA, Parfitt L, et al. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2014;190(2):208-17. http://dx.doi.org/10.1164/rccm.201403-0446OC
57. Han MK, Bach DS, Hagan PG, Yow E, Flaherty KR, Toews GB, et al. Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right-sided ventricular dysfunction. Chest. 2013;143(6):1699-708. http://dx.doi.org/10.1378/chest.12-1594
58. Fell CD. Idiopathic pulmonary fibrosis: phenotypes and comorbidities. Clin Chest Med. 2012;33(1):51-7. http://dx.doi.org/10.1016/j.ccm.2011.12.005
59. Mejia M, Carrillo G, Rojas-Serrano J, Estrada A, Suárez T, Alonso D, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136(1):10-5. http://dx.doi.org/10.1378/chest.08-2306
60. Hubbard RB, Smith C, Le Jeune I, Gribbin J, Fogarty AW. The association between idiopathic pulmonary fibrosis and vascular disease: a population-based study. Am J Respir Crit Care Med. 2008;178(12):1257-61. http://dx.doi.org/10.1164/rccm.200805-725OC
61. Dowman L, Hill CJ, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst Rev. 2014;10:CD006322. http://dx.doi.org/10.1002/14651858.cd006322.pub3
62. Ryerson CJ, Cayou C, Topp F, Hilling L, Camp PG, Wilcox PG, et al. Pulmonary rehabilitation improves long-term outcomes in interstitial lung disease: a prospective cohort study. Respir Med. 2014;108(1):203-10. http://dx.doi.org/10.1016/j.rmed.2013.11.016
63. Weill D, Benden C, Corris PA, Dark JH, Davis RD, Keshavjee S, et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2015;34(1):1-15. http://dx.doi.org/10.1016/j.healun.2014.06.014
64. Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(7):636-43. http://dx.doi.org/10.1164/rccm.200703-463PP
65. Juarez MM, Chan AL, Norris AG, Morrissey BM, Albertson TE. Acute exacerbation of
idiopathic pulmonary fibrosis-a review of current and novel pharmacotherapies. J Thorac Dis. 2015;7(3):499-519.
66. Raghu G, Brown KK, Costabel U, Cottin V, du Bois RM, Lasky JA, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178(9):948-55. http://dx.doi.org/10.1164/rccm.200709-1446OC
67. King TE Jr, Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75-81. http://dx.doi.org/10.1164/rccm.200705-732OC
68. Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR; et al. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010;181(6):604-10. http://dx.doi.org/10.1164/rccm.200906-0964OC
69. King TE Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92-9. http://dx.doi.org/10.1164/rccm.201011-1874OC
70. Malouf MA, Hopkins P, Snell G, Glanville AR; Everolimus in IPF Study Investigators. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology. 2011;16(5):776-83. http://dx.doi.org/10.1111/j.1440-1843.2011.01955.x
71. Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88-95. http://dx.doi.org/10.1164/rccm.201202-0314OC
72. Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641-9. http://dx.doi.org/10.7326/0003-4819-158-9-201305070-00003
73. Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J; MUSIC Study Group. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42(6):1622-32. http://dx.doi.org/10.1183/09031936.00104612
74. Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, Wilson EC, et al. Treating idiopathic pulmonary fibrosis with the addition of co-, , trimoxazole: a randomised controlled trial. Thorax. 2013;68(2):155-62. http://dx.doi.org/10.1136/thoraxjnl-2012-202403
75. Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pul-monary fibrosis. N Engl J Med. 2014;370(22):2093-101. http://dx.doi.org/10.1056/NEJMoa1401739











 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket