ABSTRACT
Objetivo: Avaliar o dano oxidativo (oxidação lipídica, oxidação proteica, thiobarbituric acid-reactive substances [TBARS, substâncias reativas ao ácido tiobarbitúrico], e carbonilação) e inflamação (expressão de phosphorylated AMP-activated protein kinase e de phosphorylated mammalian target of rapamycin (p-AMPK e p-mTOR, respectivamente) em tecido pulmonar e músculos do diafragma em camundongos C57BL/6 machos expostos à fumaça de cigarro (FC) por 7, 15, 30, 45 ou 60 dias. Métodos: Trinta e seis camundongos machos da espécie C57BL/6 foram divididos em seis grupos (n = 6/grupo): grupo controle e 5 grupos expostos a FC por 7, 15, 30, 45 e 60 dias, respectivamente. Resultados: Comparados aos camundongos controle, os camundongos expostos à FC apresentaram menor peso corporal em 30 dias. Nos camundongos expostos à FC (comparados aos controle) as maiores diferenças (aumentos) nos níveis de TBARS foram observados no dia 7 no músculo diafragma, comparado ao dia 45 em tecido pulmonar; as maiores diferenças (aumentos) nos níveis de carbonilas foram observados no dia 7 em ambos os tipos de tecido; e os níveis de sulfidrilas foram menores, nos dois tipos de tecidos, em todos os tempos. No tecido pulmonar e no músculo diafragma, a expressão de p-AMPK exibiu um comportamento semelhante ao dos níveis de TBARS. A expressão de p-mTOR foi maior que o valor controle nos dias 7 e 15 no tecido pulmonar, assim como no dia 45 no músculo diafragma. Conclusões: Nossos dados demonstram que a exposição à FC produz dano oxidativo tanto no tecido pulmonar quanto (primariamente) no tecido muscular, tendo um efeito adicional no músculo respiratório, como é frequentemente observado em fumantes com DPOC.
Keywords:
Oxidative stress; Mice; Respiratory system; Smoking; Inflammation.
RESUMO
Objective: To evaluate oxidative damage (lipid oxidation, protein oxidation, thiobarbituric acid-reactive substances [TBARS], and carbonylation) and inflammation (expression of phosphorylated AMP-activated protein kinase and mammalian target of rapamycin [p-AMPK and p-mTOR, respectively]) in the lung parenchyma and diaphragm muscles of male C57BL-6 mice exposed to cigarette smoke (CS) for 7, 15, 30, 45, or 60 days. Methods: Thirty-six male C57BL-6 mice were divided into six groups (n = 6/group): a control group; and five groups exposed to CS for 7, 15, 30, 45, and 60 days, respectively. Results: Compared with control mice, CS-exposed mice presented lower body weights at 30 days. In CS-exposed mice (compared with control mice), the greatest differences (increases) in TBARS levels were observed on day 7 in diaphragm-muscle, compared with day 45 in lung tissue; the greatest differences (increases) in carbonyl levels were observed on day 7 in both tissue types; and sulfhydryl levels were lower, in both tissue types, at all time points. In lung tissue and diaphragm muscle, p-AMPK expression exhibited behavior similar to that of TBARS. Expression of p-mTOR was higher than the control value on days 7 and 15 in lung tissue, as it was on day 45 in diaphragm muscle. Conclusion: Our data demonstrate that CS exposure produces oxidative damage, not only in lung tissue but also (primarily) in muscle tissue, having an additional effect on respiratory muscle, as is frequently observed in smokers with COPD.
Palavras-chave:
Estresse oxidativo; Camundongos; Sistema respiratório; Poluição por fumaça de tabaco; Inflamação
IntroductionCigarette smoke (CS) contains a large number of oxidants that have adverse effects on tissues through oxidative damage.(1,2) It is known that CS activates inflammatory cells, which can also increase polymorphonuclear cell production of oxidants in tissues, triggering oxidative stress, a crucial step in the pathogenesis of CS-induced tissue damage.(3-6) The combined effects of greater proteolytic damage, increased cell death, and decreased lung remodeling leads to emphysematous changes in the lungs.(7) Studies have shown that, in the blood of smokers,(8,9) as well as in various organs of animals chronically exposed to CS,(10) there are increases in lipid peroxidation, protein carbonylation, thiol oxidation, and DNA oxidization.
There is evidence that two central factors are involved in CS-induced direct injury or systemic inflammation: phosphorylated AMP-activated protein kinase and phosphorylated mammalian target of rapamycin (p-AMPK and p-mTOR, respectively). One recent study showed that p-AMPK activation inhibits or promotes inflammation, depending on the stimulus.(11) There is also increasing evidence that, in many cell types, an increase in intracellular reactive oxygen species (ROS) can activate p-AMPK. (12) A major integrator of environmental cues, mTOR controls cellular metabolism, growth, proliferation, and survival depending on mitogenic signals, as well as on the availability of nutrients and energy. It has now become clear that mTOR signaling plays a central role in regulating basic aspects of cell and organism behavior, and its dysregulation is strongly associated with progression of numerous human proliferative and metabolic diseases, including cancer, obesity, type 2 diabetes, and hamartoma syndrome.(13)
It is of great importance to elucidate the possible oxidative damage induced by CS directly in skeletal muscle, as well as the related structural abnormalities and the direct relationship between p-AMPK and p-mTOR, two factors associated with inflammation. Therefore, the aim of this animal study was to evaluate oxidative damage and inflammation in the lung parenchyma and diaphragm after 7, 15, 30, 45, and 60 days of exposure to CS.
MethodsIn this study, we used 36 two-month-old male C57BL/6 mice weighing 30-35 g. The animals were used and cared for in accordance with European Communities Council Directive 86/609/EEC of 24 November, 1986. The procedures adopted in this study were approved by the Research Ethics Committee of the University of Southern Santa Catarina, in the city of Criciúma, Brazil. The mice were housed in a temperature- and humidity-controlled environment (70% humidity; 20 2°C), on a 12/12-h light/dark cycle, and were given ad libitum access to water and chow (Nuvilab CR1; Nuvital Nutrientes Ltda., Colombo, Brazil). The animals were checked periodically in order to verify that they remained pathogen-free. For biochemical assays, the mice were randomized into six groups (n = 6/group): a control group; and five groups exposed to CS for 7, 15, 30, 45, and 60 days (designated CS-7, CS-15, CS-30, CS-45, and CS-60, respectively).
We used commercial filter cigarettes (MarlboroTM Red, 8 mg of tar and 0.6 mg of nicotine per cigarette; Philip Morris Products, Richmond, VA, USA).(14,15) Study animals were exposed to the smoke emitted from the burning of 12 cigarettes per day for 7, 15, 30, 45, and 60 days, as described previously by Menegali et al.(3) In brief, animals were placed in a covered inhalation chamber (40 cm long, 30 cm wide, and 25 cm high), positioned under an exhaust hood. A cigarette was coupled to a plastic 60-mL syringe so that each puff could be drawn in and subsequently expelled into the exposure chamber. One liter of smoke (20 puffs of 50 mL) was aspirated from each cigarette, each puff being immediately injected into the inhalation chamber. The animals were maintained in this smoke-air condition (3% smoke) for 6 min. We then removed the cover from the inhalation chamber and turned on the exhaust hood, which evacuated the smoke within 60 s. This process was immediately repeated. A total of four cigarettes were thus "smoked" in each treatment. The mice were subjected to these four-cigarette treatments three times per day (morning, noon, and afternoon), resulting in 72 min of CS exposure (12 cigarettes per day). (16) Each cigarette smoked produced 300 mg/m3 of total particulate matter in the exposure chamber.(3) The animals were sacrificed by cervical dislocation at 24 h after the final CS exposure. Samples of lung tissue and diaphragm muscle were homogenized in buffer solution. The homogenates were centrifuged at 1000 × g for 10 min at 4°C, and the supernatants were stored at −70°C for subsequent use in the experiments.
For histological analysis, were selected all animals in each group. The right ventricle was perfused with sterile saline (0.9%) to remove blood from the lung. The right lung was fixed (by gentle infusion of 4% phosphate buffered formalin (pH 7.2) at 25 cmH2O for 2 min through a tracheal catheter), after which it was removed and weighed. Inflated lungs were fixed for 48 h and then embedded in paraffin. Serial sagittal sections (5-µm) were obtained for histological and morphometric analyses. Macrophages and neutrophils were quantified in the alveoli. For each group, were analyzed 30 microscopic fields (10 random fields, of 26,000 mm2 each, in 3 different sections of the right lung). The number of macrophages and neutrophils (cells/mm2) were counted in a fluorescence microscope (BH-2; Olympus, Tokyo, Japan) equipped with a 40× objective.(3)
Oxidative damage was evaluated by quantifying sulfhydryl content, protein carbonyls, and malondialdehyde. Total thiol content was determined using the 5,50-dithiobis (2-nitrobenzoic acid)-DTNB-method (Sigma, St. Louis, MO, USA). The conditions of the DTNB test were as previously described.(17) In brief, 30 µL of a sample was mixed with 1 mL of PBS and 1 mM of EDTA (pH 7.5). The reaction was started by the addition of 30 µL of 10 mM DTNB stock solution in PBS. Control samples, which did not include DTNB or protein, were run simultaneously. After 30 min of incubation at room temperature, the absorbance was read at 412 nm and the amounts of 5-thio-2-nitrobenzoic acid (TNB) formed (equivalent to the amount of sulfhydryl groups) were measured. Protein carbonyls were determined using the 2,4-dinitrophenylhydrazine (DNPH) spectrophotometry method, as described by Levine et al.(18) In brief, samples containing either 2 N hydrochloric acid or DNPH were passed through columns containing Sephadex G-10 and rinsed with 2 N hydrochloric acid. The effluent was collected and mixed with guanidine hydrochloride, after which the absorbance determined at 360 nm in a spectrophotometer (SP 1105; Shanghai Spectrum Instruments Co., Ltd., Shanghai, China). The difference in absorbance with and without DNPH was calculated for all samples. Values are expressed as molar quantities using the extinction coefficient 22,000 [M-1]. Malondialdehyde, an important indicator of lipid peroxidation, was determined by spectrophotometry of the pink-colored product of thiobarbituric acid-reactive substances (TBARS). Total TBARS, as a proxy for lipid peroxidation (malondialdehyde levels), are expressed as mmol/mg of protein.(19)
Western blotting, the lung homogenates were prepared from the frozen lungs using a tissue lysis buffer (50 mM TRIS, pH 8.0, 5 mM EDTA, 150 mM NaCl, 1% nonionic detergent, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) and a protease inhibitor cocktail (Sigma). The lysates were clarified by centrifugation at 13,000 g for 15 min at 4°C; 10-30 g of protein were separated by SDS-PAGE on 10% or 15% gels; and p-AMPK and p-mTOR expression (antibodies from Cell Signaling Biotechnology, Boston, MA, USA) was analyzed by immunoblot analysis. Immunoreactivity was detected by enhanced chemiluminescence (ECL; Amersham Biosciences, Buckinghamshire, UK). The band density was determined using an imaging densitometer and analyzed with the accompanying software (GS-700 and Quantity One; Bio-Rad Laboratories, Hercules, CA, USA).(20)
Data are expressed as mean standard error of the mean. To compare means between and among groups, we used one-way ANOVA followed by Tukey's honestly significant difference post-hoc test for multiple comparisons. The level of significance was set at p < 0.05. The software used for analysis of the data was the Statistical Package for the Social Sciences, version 18.0 for Windows (SPSS Inc., Chicago, IL, USA). The sample size was based on previous studies performed in our laboratory,(3) in which similar approaches were employed.
ResultsAmong the mice evaluated in the present study, the survival rate was 100%. In comparison with the baseline values, the body weights of the animals decreased after 30, 45, and 60 days of CS exposure (27 1 vs. 23 0.8 g; p <0.01, 26 0.5 vs. 22 0.4 g; p < 0.01, and 25 0.7 vs. 20 0.3 g; p < 0.001, respectively). In addition, the body weights of the CS-60 group mice were significantly lower than were those of the control mice, as well as being significantly lower than were those of the CS-30 and CS-45 group mice (p < 0.001 for all).
In the histological analysis, lung tissue samples obtained from control mice showed thin alveolar septa and normal alveoli, whereas those obtained from mice that were exposed to CS showed destruction of the alveolar septa (starting on day 15 of exposure), alveolar enlargement, and the presence of alveolar macrophages (Figure 1A). The alveolar enlargement was significantly greater in the CS-45 group (Figure 1A). As shown in Figure 1B, the numbers of macrophages and neutrophils in the CS groups both increased significantly (in comparison with those observed for the control group) by day 7 of exposure to CS (p < 0.01). However, the difference in the number of neutrophils was more pronounced after 45 days of exposure (p < 0.001).
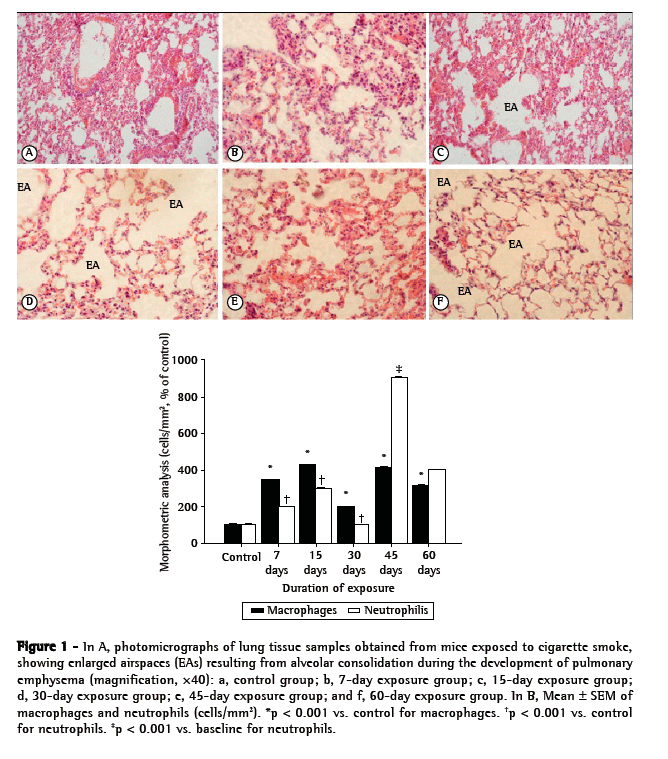
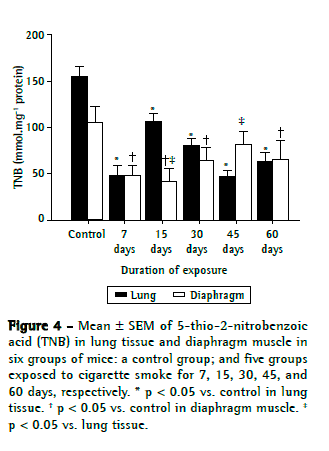
Figures 2, 3, and 4, respectively, show lipid peroxidation, protein carbonyls and sulfhydryl content in lung tissue samples and diaphragm muscle samples. In both tissue types, total TBARS increased after 7 days of exposure to CS, as did carbonyl levels. In the CS-7, CS-15, and CS-45 groups, there were differences between the lung tissue samples and diaphragm muscle samples, in terms of the degree to which carbonyl levels were increased. In the CS-15 group, the levels of TNB were significantly lower in lung tissue than in diaphragm muscle. However, by day 7 of CS exposure, TNB levels were lower than the control values in both tissue types.


The lung expression of p-AMPK was higher in the CS-15 group than in the CS-7 group. Notably, in the CS-30 and CS-45 groups, p-AMPK expression was higher in diaphragm muscle than in lung tissue (Figure 5). From day 7 of CS exposure onward, the lung expression of p-mTOR was lower in all CS-exposed groups than in the control group. However, that difference was most pronounced in the CS-7 and CS-45 groups. In the diaphragm muscle samples, p-mTOR expression began to increase by day 15 of CS, peaking by day 45 (Figure 5).
 Discussion
DiscussionIn the present study, our main objective was to characterize, at different time points, the effects induced by exposure to CS. The principal effects observed were by oxidative damage in diaphragm muscle and morphological changes in lung tissue.
The amount of neutrophils, which is associated with oxidative damage in lung tissue, was greatest on day 45 of exposure to CS. The numbers of macrophages and neutrophils are high in patients with COPD, having a direct relationship with disease severity.(21) Our data demonstrate increases in leukocytes, including macrophages and neutrophils, from day 7 to day 45 of CS exposure, which might be related to increased cell numbers and cell proliferation, resulting in immune response activation.(22) As observed, we confirmed that CS-induced pulmonary alterations appear to be the consequence of a primary inflammatory lesion characterized by the accumulation of alveolar macrophages and neutrophils in the lower respiratory tract as an immune response, which is crucial in inflammatory disease.(23) It is known that ROS play an important role in the inflammatory response to CS. Oxidative stress is characterized by higher production of ROS and decreased antioxidant levels with lipid peroxidation, thiol alterations and protein carbonylation in plasma.(24)
Pulmonary emphysema is associated with intense responses in oxidative stress, which result in a direct relationship between systemic defense activity and oxidative damage.(25,26) The oxidative damage and inflammation in lung tissue after exposure to CS have been widely studied. In addition, according to MacNee,(27) oxidative stress, as quantified by measuring plasma levels of TBARS, is associated with airflow limitation. The airflow alterations play a role in the function of respiratory muscles like the diaphragm. However, our findings demonstrate that there is an increased intensity of the inflammatory response in lung tissue starting after day 45 of exposure to CS.
According to Park et al.,(10) exposure to CS for 30 days causes significant oxidation and depletion of the glutathione pool in the lung. Those authors also concluded that the lung is a primary target of oxidative damage by cigarette smoking in the early stages, and that CS eventually exerts its oxidative effects on all organs. In our study, it was observed that CS-induced oxidative damage caused changes not only in the lungs but also in the diaphragm. We found that exposure to CS for 30-45 days was sufficient to generate higher levels of oxidative damage in skeletal muscle (the diaphragm).
A recent study showed that the main limitation found in COPD patients might be related to the mechanism of slow cardiac output associated with airflow limitation.(28) Chiappa et al.(29) tested conditions that improve oxygen delivery and uptake as strategies in COPD patients. The authors demonstrated that one such strategy-the use of heliox (a mixture of 79% helium and 21% oxygen)-is able to ameliorate expiratory flow limitation and dynamic hyperinflation, accelerating the dynamics of peripheral muscle utilization of oxygen as a consequence of improved delivery during high-intensity exercise in patients with moderate to severe COPD. We believed that these interactions might be linked with redox balance and inflammatory responses. One recent study suggested that, in the clinical management of acute lung injury, the use of heliox has the combined therapeutic benefits of reducing mechanical and oxidative stress, thus attenuating lung inflammation.(30)
Oxidative damage generated by exposure to CS in skeletal muscle can lead to loss of muscle function, manifesting as a loss of muscle strength and a consequent higher susceptibility to fatigue. (1,31) The present investigation is the first to provide evidence of oxidative changes induced by ROS in diaphragm muscle proteins in animals chronically exposed to CS. We found that protein oxidation was significantly increased in the diaphragm after 7 days of exposure to CS. The carbonylation of the diaphragm was highest after 30-45 days of exposure, as opposed to carbonylation in the lung, which did not peak until day 60. Our data indicate that exposure to CS primarily affects the diaphragm, which can translate to a significant loss of locomotor and respiratory muscle function in pulmonary emphysema.
According to Barreiro et al.,(1) the effects of smoking-induced muscle protein oxidation appear at an earlier stage in the quadriceps muscle than in the respiratory muscles. These findings underscore the concept that CS per se is likely to be involved in direct tissue toxicity in the skeletal muscles of CS-exposed mice, regardless of lung and bronchial alterations. In addition, we observed that the same animals acutely exposed to CS exhibited a significant increase in TBARS, together with a reduction in muscle levels of sulfhydryl, immediately after exposure. Carbonylation is crucial to triggering activation of the oxidative pathway and promoting lipid peroxidation.
In this animal study of chronic CS exposure, we have shown that pulmonary function decreases in parallel with the duration of exposure, similar to what has been observed in humans.(32) In addition, chronic CS exposure has been shown to cause airflow obstruction.(33) When we analyzed the expression of p-AMPK and p-mTOR in lung tissue, we observed decreased expression of p-mTOR, a result that was expected because p-mTOR expression is associated with cell metabolism, growth, proliferation, and survival, depending on mitogenic signals, as well as on the availability of nutrients and energy.
The increased expression of p-mTOR observed in the diaphragm from day 15 to day 45 of CS exposure can be explained by the possible increase in muscle protein synthesis related to a state of physiological stress.(34) In a rat model of CS exposure, Kozma et al.(5) demonstrated that airway resistance and respiratory system resistance were higher in exposed animals than in unexposed animals. This increase in airway resistance might result in a greater diaphragmatic work, which would explain the increased diaphragm expression of p-mTOR in our CS-15, CS-30, and CS-45 groups, given that p-mTOR expression is known to be elevated in situations of muscle hypertrophy. (35) In our CS-60 group, there was a significant reduction in p-mTOR expression, which was an expected result, because myopathy is associated with reduced expression of p-mTOR.(36) Such myopathy is common in chronic lung diseases.(1) However, in our study, the expression of p-AMPK was increased only from day 30 to day 45 of CS exposure. This fact might be explained by the fact that the increased p-AMPK expression was accompanied by an increase in oxidative stress, which is clear when we look at the increase in carbonyl by day 30 of CS exposure. Increasing evidence suggests that p-AMPK can be activated by an increase in intracellular ROS in many cell types.(12) Accordingly, whether the ROS-sensitive p-AMPK signaling pathway is involved in toxic smoke-induced lung inflammation remains to be investigated.
Perang et al.(37) were the first to report a detailed AMPK signaling pathway responsible for inducing interleukin (IL)-8 expression by toxic smoke exposure in lung epithelial cells. In this pathway, increased intracellular levels of ROS level constitute the vital trigger, because removal of intracellular ROS by N-acetyl-cysteine reduced the activation of AMPK, c-Jun N-terminal kinase, and extracellular signal-regulated kinase, as well as the induction of IL-8.(37) Previous studies have reported that toxic smoke can increase the intracellular ROS level in lung cells, although the mechanism remains unclear.(38)
In conclusion, our study shows, for the first time, that oxidative alterations in muscle proteins occur in the diaphragm as early as day 7 days of exposure to CS. In addition, this event occurred concomitantly with the parenchymal abnormalities induced by CS in the lungs, suggesting a direct toxic effect of CS on skeletal muscle proteins. However, our data also make it more obvious that pulmonary emphysema is a complex disease that has a negative impact on the whole body. Furthermore, we found that the oxidative damage caused by CS exposure occurs first in skeletal muscle and then in lung tissue.
AcknowledgementsWe are grateful to our colleagues in the Laboratory of Exercise Biochemistry and Physiology at the University of Southern Santa Catarina, in Criciuma, Brazil, for their collaboration.
References1. Barreiro E, Peinado VI, Galdiz JB, Ferrer E, Marin-Corral J, Sánchez F, et al. Cigarette smoke-induced oxidative stress: A role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am J Respir Crit Care Med. 2010;182(4):477-88. http://dx.doi.org/10.1164/rccm.200908-1220OC
2. Fusco L, Pêgo-Fernandes P, Xavier A, Pazetti R, Rivero D, Capelozzi V, et al. Modelo experimental de enfisema pulmonar em ratos induzido por papaína. J Pneumol. 2002;28:1-7.
3. Menegali BT, Nesi RT, Souza PS, Silva LA, Silveira PC, Valença SS, et al. The effects of physical exercise on the cigarette smoke-induced pulmonary oxidative response. Pulm Pharmacol Ther. 2009;22(6):567-73. http://dx.doi.org/10.1016/j.pupt.2009.08.003
4. Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87(3):1047-82. http://dx.doi.org/10.1152/physrev.00048.2006
5. Kozma Rde L, Alves EM, Barbosa-de-Oliveira VA, Lopes FD, Guardia RC, Buzo HV, et al. A new experimental model of cigarette smoke-induced emphysema in Wistar rats. J Bras Pneumol. 2014;40(1):46-54. http://dx.doi.org/10.1590/S1806-37132014000100007
6. Valença SS, Porto LC. Immunohistochemical study of lung remodeling in mice exposed to cigarette smoke. J Bras Pneumol. 2008;34(10):787-95. http://dx.doi.org/10.1590/S1806-37132008001000006
7. Tuder RM, Petrache I, Elias JA, Voelkel NF, Henson PM. Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol. 2003;28(5):551-4. http://dx.doi.org/10.1165/rcmb.F269
8. Kalra J, Chaudhary AK, Prasad K. Increased production of oxygen free radicals in cigarette smokers. Int J Exp Pathol. 1991;72(1):1-7.
9. Frei B, Forte TM, Ames BN, Cross CE. Gas phase oxidants of cigarette smoke induce lipid peroxidation and changes in lipoprotein properties in human blood plasma. Protective effects of ascorbic acid. Biochem J. 1991;277(Pt 1):133-8.
10. Park EM, Park YM, Gwak YS. Oxidative damage in tissues of rats exposed to cigarette smoke. Free Radic Biol Med. 1998;25(1):79-86. http://dx.doi.org/10.1016/S0891-5849(98)00041-0
11. Chang MY, Ho FM, Wang JS, Kang HC, Chang Y, Ye ZX, et al. AICAR induces cyclooxygenase-2 expression through AMP-activated protein kinase-transforming growth factor-beta-activated kinase 1-p38 mitogen-activated protein kinase signaling pathway. Biochem Pharmacol. 2010;80(8):1210-20. http://dx.doi.org/10.1016/j.bcp.2010.06.049
12. Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010;285(43):33154-64. http://dx.doi.org/10.1074/jbc.M110.143685
13. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274-93. http://dx.doi.org/10.1016/j.cell.2012.03.017
14. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco smoke and involuntary smoking. IARC Monogr Eval Carcinog Risks Hum. 2004;83:1-1438.
15. Counts ME, Morton MJ, Laffoon SW, Cox RH, Lipowicz PJ. Smoke composition and predicting relationships for international commercial cigarettes smoked with three machine-smoking conditions. Regul Toxicol Pharmacol. 2005;41(3):185-227. http://dx.doi.org/10.1016/j.yrtph.2004.12.002
16. Valenca SS, Bezerra FS, Romana-Souza B, Paiva RO, Costa AM, Porto LC. Supplementation with vitamins C and E improves mouse lung repair. J Nutr Biochem. 2008;19(9):604-11. http://dx.doi.org/10.1016/j.jnutbio.2007.08.004
17. Aksenov MY, Markesbery WR. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer's disease. Neurosci Lett. 2001;302(2-3):141-5. http://dx.doi.org/10.1016/S0304-3940(01)01636-6
18. Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, et al. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464-78. http://dx.doi.org/10.1016/0076-6879(90)86141-H
19. Draper HH, Hadley M. Malondialdehyde Determination as Index of Lipid-Peroxidation. Methods Enzymol. 1990;186:421-31. http://dx.doi.org/10.1016/0076-6879(90)86135-I
20. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350-4.
21. Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC, et al. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med. 2001;164(3):469-73. http://dx.doi.org/10.1164/ajrccm.164.3.2007149
22. Duong C, Seow HJ, Bozinovski S, Crack PJ, Anderson GP, Vlahos R. Glutathione peroxidase-1 protects against cigarette smoke-induced lung inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2010;299(3):L425-33. http://dx.doi.org/10.1152/ajplung.00038.2010
23. Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A, et al. Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L931-45. http://dx.doi.org/10.1152/ajplung.00201.2005
24. Park EM, Park YM, Gwak YS. Oxidative damage in tissues of rats exposed to cigarette smoke. Free Radic Biol Med. 1998;25(1):79-86. http://dx.doi.org/10.1016/S0891-5849(98)00041-0
25. Kluchová Z, Petrásová D, Joppa P, Dorková Z, Tkácová R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol Res. 2007;56(1):51-6.
26. Pinho RA, Chiesa D, Mezzomo KM, Andrades ME, Bonatto F, Gelain D, et al. Oxidative stress in chronic obstructive pulmonary disease patients submitted to a rehabilitation program. Respir Med. 2007;101(8):1830-5. http://dx.doi.org/10.1016/j.rmed.2007.02.004
27. MacNee W. Oxidative stress and lung inflammation in airways disease. Eur J Pharmacol. 2001;429(1-3):195-207. http://dx.doi.org/10.1016/S0014-2999(01)01320-6
28. Chiappa GR, Borghi-Silva A, Ferreira LF, Carrascosa C, Oliveira CC, Maia J, et al. Kinetics of muscle deoxygenation are accelerated at the onset of heavy-intensity exercise in patients with COPD: relationship to central cardiovascular dynamics. J Appl Physiol (1985). 2008;104(5):1341-50. http://dx.doi.org/10.1152/japplphysiol.01364.2007
29. Chiappa GR, Queiroga F Jr, Meda E, Ferreira LF, Diefenthaeler F, Nunes M, et al. Heliox improves oxygen delivery and utilization during dynamic exercise in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009 Jun 1;179(11):1004-10. http://dx.doi.org/10.1164/rccm.200811-1793OC
30. Nawab US, Touch SM, Irwin-Sherman T, Blackson TJ, Greenspan JS, Zhu G, et al. Heliox attenuates lung inflammation and structural alterations in acute lung injury. Pediatr Pulmonol. 2005;40(6):524-32. http://dx.doi.org/10.1002/ppul.20304
31. Barreiro E, Rabinovich R, Marin-Corral J, Barberà JA, Gea J, Roca J. Chronic endurance exercise induces quadriceps nitrosative stress in patients with severe COPD. Thorax. 2009;64(1):13-9. http://dx.doi.org/10.1136/thx.2008.105163
32. Gold DR, Wang X, Wypij D, Speizer FE, Ware JH, Dockery DW. Effects of cigarette smoking on lung function in adolescent boys and girls. N Engl J Med. 1996;335(13):931-7. http://dx.doi.org/10.1056/NEJM199609263351304
33. Wright JL, Sun JP, Vedal S. A longitudinal analysis of pulmonary function in rats during a 12 month cigarette smoke exposure. Eur Respir J. 1997;10(5):1115-9.
34. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471-84. http://dx.doi.org/10.1016/j.cell.2006.01.016
35. Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19-37. http://dx.doi.org/10.1146/annurev.biochem.75.103004.142622
36. Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol. 2009;187(6):859-74. http://dx.doi.org/10.1083/jcb.200903131
37. Perng DW, Chang TM, Wang JY, Lee CC, Lu SH, Shyue SK, et al. Inflammatory role of AMP-activated protein kinase signaling in an experimental model of toxic smoke inhalation injury. Crit Care Med. 2013;41(1):120-32. http://dx.doi.org/10.1097/CCM.0b013e318265f653
38. Lee TS, Liu YJ, Tang GJ, Yien HW, Wu YL, Kou YR. Wood smoke extract promotes both apoptosis and proliferation in rat alveolar epithelial type II cells: the role of oxidative stress and heme oxygenase-1. Crit Care Med. 2008;36(9):2597-606. http://dx.doi.org/10.1097/CCM.0b013e318184979c
Study carried out in the Laboratory for Research in the Physiopathology of Exercise, Porto Alegre Hospital de Clínicas, Porto Alegre, Brazil.
Correspondence to: Gaspar Rogério da Silva Chiappa. Laboratório de Fisiopatologia do Exercício, Hospital de Clinicas de Porto Alegre, Rua Ramiro Barcelos, 2350, sala 2061, CEP 90035-903, Porto Alegre, RS, Brasil.
Tel. 55 51 3359 6332. E-mail: gaspar.chiappa@gmail.com
Financial support: This study received financial support from the Brazilian Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, National Council for Scientific and Technological Development), the Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (FAPESC, Foundation for the Support of Research and Innovation in the state of Santa Catarina), and the Universidade do Extremo Sul de Santa Catarina (UNESC, University of Southern Santa Catarina).
Submitted: 14 March 2014. Accepted, after review: 2 July 2014.
**A versão completa em português deste artigo está disponível em www.jornaldepneumologia.com.br
About the authorsSamanta Portão de Carlos1
Student. Department of Physical Therapy. University of Southern Santa Catarina, Criciúma, Brazil.
Alexandre Simões Dias
Professor. Graduate Program in Movement Sciences and Pulmonology Sciences, Federal University of Rio Grande do Sul, Porto Alegre, Brazil.
Luiz Alberto Forgiarini Júnior
Professor. Methodist University, Instituto Porto Alegre (IPA, Porto Alegre Institute), Porto Alegre, Brazil.
Patrícia Damiani Patricio
Student Department of Physical Therapy. University of Southern Santa Catarina, Criciúma, Brazil.
Thaise Graciano
Student. Department of Physical Therapy. University of Southern Santa Catarina, Criciúma, Brazil.
Renata Tiscoski Nesi
Professor. Institute of Biomedical Science, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil.
Samuel Valença
Professor. Laboratory for Research in the Physiopathology of Exercise, Department of Cardiology, Porto Alegre Hospital de Clínicas, Porto Alegre, Brazil.
Adriana Meira Guntzel Chiappa
Physical Therapist. Intensive Care Unit, Porto Alegre Hospital de Clínicas, Porto Alegre, Brazil.
Gerson Cipriano Jr
Professor. Health Sciences and Technologies Program, Department of Physical Therapy, University of Brasília, Brasília, Brazil.
Claudio Teodoro de Souza
Professor. Department of Physical Therapy. University of Southern Santa Catarina, Criciúma, Brazil.
Gaspar Rogério da Silva Chiappa
Professor. Laboratory for Research in the Physiopathology of Exercise, Department of Cardiology, Porto Alegre Hospital de Clínicas, Porto Alegre, Brazil; and Epidemiology and Public Health Research Group, Serra Gaucha College, Caxias do Sul, Brazil.











 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket