ABSTRACT
In patients with myositis, the lung is commonly involved, and the presence of anti-aminoacyl-tRNA synthetase (anti-ARS) antibodies marks the presence or predicts the development of interstitial lung disease (ILD). A distinct clinical entity-antisynthetase syndrome-is characterized by the presence of anti-ARS antibodies, myositis, ILD, fever, arthritis, Raynaud's phenomenon, and mechanic's hands. The most common anti-ARS antibody is anti-Jo-1. More recently described anti-ARS antibodies might confer a phenotype that is distinct from that of anti-Jo-1-positive patients and is characterized by a lower incidence of myositis and a higher incidence of ILD. Among patients with antisynthetase syndrome-related ILD, the response to immunosuppressive medications is generally, but not universally, favorable.
Keywords:
Lung diseases, interstitial; Pneumonia; Infection.
RESUMO
Em pacientes com miosite, é comum o comprometimento pulmonar, e a presença de anticorpos anti-aminoacil-RNAt sintetase (anti-ARS) é preditora da presença ou do desenvolvimento de doença pulmonar intersticial (DPI). Uma entidade clínica distinta - a síndrome antissintetase - é caracterizada pela presença de anticorpos anti-ARS, miosite, DPI, artrite, fenômeno de Raynaud e "mãos de mecânico". O mais comum anticorpo anti-ARS é o anti-Jo-1. Anticorpos anti-ARS mais recentemente descritos podem conferir um fenótipo que é distinto daquele de pacientes com positividade para anti-Jo-1, sendo caracterizado por uma menor incidência de miosite e uma maior incidência de DPI. Nos pacientes com DPI relacionada à síndrome antissintetase, a resposta a medicações imunossupressoras é em geral favorável.
Palavras-chave:
Pneumonias intersticiais idiopáticas; Pneumonia; Infecção.
IntroductionThe idiopathic inflammatory myopathies (IIMs) are a heterogeneous group of acquired muscle diseases characterized by varying types and degrees of skeletal muscle inflammation. Three major subtypes are recognized: sporadic inclusion-body myositis; polymyositis (PM); and dermatomyositis (DM). However, aside from the skin involvement, PM and DM are similar enough that authors often use the term "PM/DM" when making reference to them.
Although PM and DM both typically manifest as progressive skeletal muscle weakness that spares the face and eyes, manifestations not involving the skeletal muscles are common and can be more clinically significant than the myositis. For example, abnormalities of the swallowing mechanism, cardiac involvement, and pulmonary disease are all frequently found in patients with PM/DM. In fact, the direct or indirect pulmonary manifestations of PM/DM are a major cause of morbidity and mortality. In 5% of PM/DM patients, respiratory muscle weakness leads to hypoventilation, resulting in atelectasis and complicating pneumonia.(1) A potentially fatal condition, aspiration pneumonia secondary to pharyngeal muscle dysfunction, occurs in 17% of patients with PM/DM.(2) Interstitial lung disease (ILD) is a long recognized complication, having first been described in the 1950s.(3) Mainly due to the sensitivity of chest CT, ILD is now recognized as the most common non-musculoskeletal manifestation of the disease; from one half to three quarters of PM/DM patients have evidence of ILD on HRCT scans of the chest.(4,5)
Autoantibodies are detectable in the sera of 50% of PM/DM patients and consist of myositis-associated and myositis-specific antibodies (MAAs and MSAs, respectively).(6) The MAAs are not specific to PM/DM and are found in a variety of autoimmune diseases. The MSAs are divided into those directed at the following: components of a nucleosome remodeling complex (anti-Mi-2)(7); a macromolecular complex involved in RNA degradation and processing (anti-PM/Scl)(8); ribonucleoproteins involved in translational transport (anti-signal recognition particle, or anti-SRP); and ribonucleoproteins involved in protein synthesis (anti-aminoacyl-tRNA synthetase antibodies, also known as antisynthetase antibodies, or anti-ARS).(9)
A specific subset of PM/DM patients have a clinical syndrome consisting of the presence of anti-ARS antibodies, ILD, and some of the following clinical features: fever, arthralgias, Raynaud's phenomenon, and exanthema on the hands (also referred to as mechanic's hands). This combination of findings is designated antisynthetase (AS) syndrome. In this paper, we review the data on PM/DM-related ILD, with a particular focus on AS syndrome.
AS syndromeHistoryThe association of PM/DM and extraskeletal manifestations has been recognized since the 1950s,(3) although it was not until the 1990s that AS syndrome was defined as a unique clinical entity. In 1990, Marguerie et al. described a series of 29 subjects with PM/DM and additional clinical features, including Raynaud's phenomenon, inflammatory arthritis, ILD, and a handful of anti-ARS antibodies (e.g., anti-Jo-1, PL-7, or PL-12).(10)
In a subsequent study, Love et al. built on these findings by analyzing a cohort of PM/DM patients stratified by autoantibody profile.(11) The authors recognized significant differences in signs, symptoms, immunogenetics, and prognosis among the subgroups. In particular, PM/DM patients with anti-ARS antibodies were more likely to have fever, dyspnea, mechanic's hands, arthritis, and ILD than were those without such antibodies.
AutoantibodiesAlthough MAAs are common, they are not universally seen in PM/DM patients; antinuclear antibodies (ANAs), anti-SSA/Ro antibodies, and anti-U1 ribonucleoprotein (anti-U1-RNP) antibodies are found in 52%, 12%, and 11%, respectively.(11) In contrast, MSAs appear to define specific clinical phenotypes. Anti-Mi-2 antibodies are found in 4-14% of PM/DM patients and are associated with diffuse, cutaneous, steroid-sensitive skin involvement.(7,9,12) Anti-PM/Scl antibodies are found in approximately 8% of the patients who have the PM/systemic sclerosis overlap phenotype, which typically consists of skin manifestations of systemic sclerosis, together with clinical features similar to those seen in patients with anti-ARS antibodies.(13,14) Anti-SRP autoantibodies are present in 4% of the patients with myositis and might portend a poor prognosis, given their apparent association with severe muscle disease and with cardiac involvement that is poorly responsive to treatment.(9,15,16)
Anti-ARS antibodies are directed against cytoplasmic enzymes that catalyze the formation of the aminoacyl-tRNA complex from an amino acid and its cognate tRNA. To date, eight different anti-ARS antibodies have been described: anti-PL-7 (anti-threonyl)(17); anti-PL-12 (anti-alanyl)(18); anti-OJ (anti-isoleucyl)(19); anti-EJ (anti-glycyl)(19); anti-KS (anti-asparaginyl)(20); anti-ZO (anti-phenylalanyl)(21); anti-tyrosyl(22); and anti-Jo-1 (anti-histidyl).(23) All of these antibodies are directed at functionally related enzymes and are mutually exclusive in a given patient. Although initially believed to represent the general presence of an autoimmune myositis, it has subsequently become clear that they are in fact markers of AS syndrome clinical phenotypes.(10)
Anti-Jo-1 was the first anti-ARS to be discovered and characterized.(23) Perhaps because the other anti-ARS antibodies have only more recently been identified and few laboratories have the ability to test for them, anti-Jo-1 is the most commonly identified anti-ARS antibody, and most of the clinical data about AS syndrome is based on patients who are anti-Jo-1-positive. It is found in 20-30% of PM patients, in 5-10% of those with DM,(11) and in 75% of all reported cases in which an anti-ARS is present. The strongest predictor of ILD in PM/DM is the presence of anti-Jo-1-over 70% of anti-Jo-1-positive patients have ILD(4,24)-and disease activity might correlate with the levels of the antibody.(25) The significance of a positive result for anti-Jo-1 in the setting of ILD without any other criterion for AS syndrome remains unclear. Other anti-ARS antibodies are far less common: anti-PL-7 or anti-PL-12 are detected in 2-5% of the patients; and the rest (anti-OJ, anti-EJ, anti-KS, anti-ZO, and anti-tyrosyl) are identified in < 2% of the patients.(26)
EpidemiologyAlthough the overall incidence of inflammatory myopathies ranges from 6 to 10 per 1,000,000 population,(1,27) the incidence of anti-Jo-1 positivity ranges from 1.2 to 2.5 per million, with a reported prevalence of 1.5 per 100,000 population.(28,29) The average age at diagnosis of patients with anti-Jo-1-positive AS syndrome is 50 years (range, 22-74 years).(10,30) A predominantly female condition, the mean female/male ratio of AS syndrome is 2:1 (in some studies, this ratio is as great as 13:1).(31) In Japan and in the USA, patients with anti-Jo-1-positive AS syndrome appear to have similar phenotypes.(32) In one study of anti-Jo-1-positive patients, ILD was found to be more severe in African-Americans than in Whites.(30) There is an association between certain HLAs and PM: among anti-Jo-1-positive patients, 91% are positive for HLA-DR3, and 80% are positive for HLA-DQ2.(11,33)
Clinical characteristics of AS syndromeThe population of patients with AS syndrome present with a constellation
of clinical and biochemical features. Key features for diagnosis include the presence of an anti-ARS antibody, accompanied by myositis, ILD, or both. Although arthritis, mechanic's hands, and Raynaud's phenomenon support the diagnosis, their presence is not necessary. In Chart 1, we propose diagnostic criteria for AS syndrome.
 Myositis
MyositisMost anti-Jo-1-positive patients have PM, a smaller proportion having DM or overlap syndromes (57% vs. 28% in one study).(31) Muscle histopathology in anti-Jo-1-positive patients differs from that observed in antibody-negative patients. In contrast to antibody-negative patients, in whom there is predominately endomysial and perivascular inflammation, anti-Jo-1-positive patients have fragmentation of the perimysial connective tissue with macrophage predominant inflammation and, in rare cases, vascular involvement.(34)
Myositis is not universal and can develop subsequent to the diagnosis of AS syndrome. Biochemical myositis is recognized to precede ILD in 12% of the patients with AS syndrome, whereas ILD precedes myositis in 37% of these, and their onset is simultaneous in 50%.(35) In one recent study, 31% of the patients who presented with ILD and anti-Jo-1 antibodies met the criteria for a diagnosis of myositis at the outset, and 56% of the patients eventually developed myositis over a median follow-up period of 62 months.(36) Because the diagnosis of AS syndrome can occur in the absence of myositis (termed "amyopathic" AS syndrome), the true incidence of clinically significant myositis in anti-Jo-1-positive patients with AS syndrome is unknown-most case series have required an a priori diagnosis of myositis.
Arthritis and other featuresIn anti-Jo-1-positive AS syndrome, arthritis, which is observed in up to 75% of the patients,(24,31,37) is symmetrical, involving the wrists and metacarpal/phalangeal joints, whereas the proximal interphalangeal joints, shoulders, knees, and elbows are less affected. The majority of articular symptoms occur early in the disease, are usually mild, and resolve with the treatment of myositis. In the majority of the patients with AS syndrome, such symptoms are non-deforming; however, up to a third of the patients will have joint subluxation without erosion and occasional periarticular calcinosis.(38) Joint effusions with inflammatory synovial fluid can occur.(39) Raynaud's phenomenon is often associated with the presence of anti-Jo-1 antibodies,(24,31,37) but mechanic's hands are a rare finding.(31,37) Fever is reported to occur in up to 35% of patients at some point during the course of disease.(31,35)
Associated diseasesThe association between inflammatory myopathies (DM in particular) and malignancy is well recognized. Oddly, anti-ARS antibodies seem to be somewhat protective-subjects with anti-ARS antibodies are less likely to have malignancy.(40,41) However, two cases of malignancy (one of poorly differentiated lung adenocarcinoma and one of colon cancer) have been reported in patients with DM, positivity for anti-Jo-1 antibodies, and features of AS syndrome.(42,43) There have been multiple case reports of patients with AS syndrome developing other clinical disorders, including sarcoidosis,(44) myasthenia gravis,(45) ankylosing spondylitis,(46)
Klinefelter syndrome,(47) and Kennedy's disease (an X-linked neuromuscular disease).(48) One case of suspected drug-induced AS syndrome has also been described.(49)
ILDDepending upon the inclusion criteria, the method of investigation, and the length of the follow-up period, up to 75% of PM/DM patients will have some evidence of ILD at the time of diagnosis of the connective tissue disease. Therefore, in patients with PM/DM and respiratory symptoms, ILD should always be included in the differential diagnosis.
Clinical presentationIn many patients with AS syndrome-related ILD, the onset of dyspnea is gradual (occurring over a matter of months). However, in a subset of patients, the onset of ILD, fever, and respiratory insufficiency is abrupt (occurring over a few days or weeks).(36,50,51) Among a recently described group of 32 subjects with AS syndrome, equal numbers presented with acute respiratory insufficiency and a more insidious onset of dyspnea.(36)
Physiological, chest imaging, BAL fluid, and histological findingsPatients with AS syndrome-related ILD present with a restrictive pulmonary pattern and impaired gas exchange (mean TLC ≤ 60% of predicted; mean FVC ≤ 60% of predicted, and mean DLCO ≤ 50% of predicted).(10,30,36)An obstructive pattern is rare.
A pattern of nonspecific interstitial pneumonia (NSIP), with or without areas of consolidation (suggestive of organizing pneumonia), is usually revealed on HRCT scans. A usual interstitial pneumonia (UIP) pattern can also be seen.(30,36) In our experience, many patients present with a distinct pattern that is highly suggestive of AS syndrome-related ILD. In this pattern, there are (predominantly basilar) reticular and ground-glass opacities, accompanied by a loss of lung volume, traction bronchiectasis, and scattered (usually peribronchovascular) areas of consolidation (Figure 1).(52)
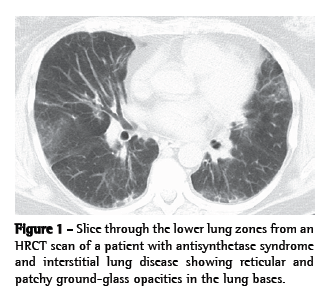
In BAL fluid, the few data available suggest that lymphocytes (primarily CD8-positive cells) predominate.(10,35,36,53) On surgical lung biopsy, an NSIP pattern is the most common; a UIP pattern, organizing pneumonia, or diffuse alveolar damage are observed in less than 20% of the patients (Figure 2).(10,35,36,53) In our experience, the combination of an NSIP pattern and organizing pneumonia is more common than is either in isolation.
 Prognosis
PrognosisRecognizing the limitations imposed by the variability in design, the retrospective format, and the limited number of the studies available, we can nevertheless surmise that the usual course of ILD in patients with AS syndrome is stabilization or improvement. Among patients who have ILD in the setting of PM/DM, the presence or absence of an anti-ARS antibody does not seem to affect the long-term outcome, although the antibodies might be markers of the patients who are more likely to have "flares" of ILD that require courses of high-dose corticosteroids or of other
immunosuppressants.(54)
Anti-Jo-1-negative AS syndromeRecent data suggest that there are subtypes of AS syndrome whose clinical characteristics (myositis, skin disease, severe ILD, or other clinical features, such as esophageal involvement or pulmonary hypertension) depend on which specific anti-ARS antibody is present (Chart 2).(5,10,30-32,35-38,53,55-60) For example, among patients with amyopathic AS syndrome, anti-Jo-1 is detected far less frequently than are other anti-ARS antibodies.(61)
 Anti-PL-12
Anti-PL-12There have been six case series comprising 69 subjects that have reported the clinical syndrome associated with anti-PL-12 antibodies.(10,18,52,62-64) Similarly to patients with anti-Jo-1 antibodies, the average age of those patients at the time of diagnosis was 52 years (range, 22-87 years). Over 75% of the subjects with anti-PL-12 antibodies were women, and there was a high incidence of MAAs (over 80% of the subjects in one study), including anti-U1-RNP and anti-Sm antibodies, which are rarely seen in patients with anti-Jo-1-positive AS syndrome.(37,64) Compared with anti-Jo-1-positive AS syndrome patients, those testing positive for anti-PL-12 antibodies have a higher incidence of ILD (70-100%) and a lower incidence of biochemical myositis. Although early studies reported an incidence of myositis of 60-100%,(10,18,64) more recent reports have identified myositis in a smaller proportion of subjects (0-50%).(52,62,63) In a case series of subjects with amyopathic AS syndrome and ILD, anti-PL-12 antibodies were identified in 60% of the subjects.(65,66)
Among anti-PL-12-positive patients, Raynaud's phenomenon occurs in 40-100% but mechanic's hands are rare. Data from some studies suggest a higher incidence of pulmonary hypertension and esophageal involvement.(62,63,67) A UIP pattern of lung injury might be more common in patients with anti-PL-12-positive AS syndrome than in those with the anti-Jo-1-positive form, which was present in over half of the subjects for whom histological data were available.(63) Prognosis seems to depend on the severity of ILD, and the response to immunosuppressive therapy is variable.(52)
Anti-PL-7In six published studies, a combined total of 21 anti-PL-7-positive subjects were evaluated, and 63% were women.(10,17,52,68,69) The average age at diagnosis was in the fourth decade of life (range, 15-68 years).
Compared with anti-Jo-1-positive AS syndrome patients, those testing positive for anti-PL-7 antibodies appear to have a higher incidence of ILD and a lower incidence of myositis. In a study of ANA-negative and anti-Jo-1-negative AS syndrome subjects with ILD, anti-PL-7 antibodies were identified in 77%, 42% of whom had muscle involvement.(10,52) Raynaud's phenomenon and arthritis have been reported.
Other anti-ARS antibodiesThere have been isolated case reports of AS syndrome in subjects testing positive for anti-ARS antibodies that were discovered more recently. A study conducted in Japan evaluated 7 subjects with anti-OJ antibodies(70): 4 were women; 4 had myositis; and all had ILD. In other studies, a total of 6 subjects with anti-KS antibodies have been described: all were women; none had myositis; but all had ILD.(20,35,71) Among 4 such subjects who underwent surgical lung biopsy, a UIP-pattern was identified in 2, and a NSIP-pattern was identified in 2. There has been only one reported case of anti-ZO-positive AS syndrome: a 49-year-old woman with myositis, arthritis, Raynaud's phenomenon, and an HRCT pattern consistent with NSIP.(21)
Therapy and prognosisEarly reports looking at IIM-associated ILD suggested a favorable response to therapy with prednisone(72,73): 30-40% of the subjects improved; and 20-40% stabilized based on symptoms and pulmonary function. A subset of patients are resistant to corticosteroids, and there are reports of remission induced by the addition of azathioprine, methotrexate, cyclophosphamide, cyclosporine, tacrolimus, or mycophenolate mofetil.(74-80) In early studies, deaths from ILD were rare: the death rate from respiratory failure was reported to be approximately 10% at a median follow-up period of 4 years. Among all comers (i.e., with or without AS syndrome), the five-year survival rate for individuals with PM/DM-related ILD appears to be similar to that reported for those with idiopathic NSIP-60% for both.(74)
In a comprehensive evaluation of patients with IIM and ILD followed for a median duration of 53 months, one group of authors observed that among the subjects treated with immunosuppressive drugs, ILD resolved in 19% and improved in 55%.(24) The authors reported one-, three-, and five-year survival rates of 94.4%, 90.4%, and 86.5%, respectively. In 25%, ILD progressed, and, in that group, there was a higher incidence of neutrophilia in BAL fluid and a UIP pattern on biopsy. As observed by other investigators, the presence of anti-Jo-1 was not associated with the outcome.(54,57) One group of investigators prospectively followed 20 consecutive patients with ILD and PM/DM, the majority of whom had AS syndrome: 10 had a stable, nonprogressive course, and 10 had rapidly progressive ILD. All of those with progressive disease stabilized or improved with the addition of cyclophosphamide (to the background therapy with prednisolone), although relapses occurred.(76) In one study focusing on patients with AS syndrome, the majority of the subjects improved with immunosuppression: 72% of the patients stabilized with the treatment. However, 28% developed progressive respiratory failure and died. Relapse was more common in those treated with corticosteroids alone. There were no differences in the response to therapy or in the outcome at one year among those presenting with acute respiratory failure or gradual onset of dyspnea.(36) Another group of authors studied 14 anti-ARS-positive ILD patients and observed that ILD improved with corticosteroids (with or without cyclosporine) in 64%; only 1 patient died from respiratory failure.(35) One group of investigators observed that, at a mean disease duration of 5 years, the subjects with anti-ARS antibodies had a mortality rate of 21% compared with that of 7% in the subjects with no MSA.(11) The poorer outcome in these subjects has been attributed to the presence of ILD.(81,82)
Our usual approach to therapy for PM/DM-related ILD (including patients with AS syndrome) is to use a combination of glucocorticoids and a steroid-sparing agent (usually mycophenolate mofetil or azathioprine). We use cyclophosphamide for cases with clinically severe or rapidly progressive ILD. For patients in whom ILD worsens despite aggressive conventional therapy, rituximab has been used with moderate success,(83,84) with the stabilization or improvement of 7 of the 11 patients evaluated in one case series (Chart 3).(85)
 References
References1. Fathi M, Lundberg IE, Tornling G. Pulmonary complications of polymyositis and dermatomyositis. Semin Respir Crit Care Med. 2007;28(4):451-8.
2. Marie I, Hachulla E, Chérin P, Hellot MF, Herson S, Levesque H, et al. Opportunistic infections in polymyositis and dermatomyositis. Arthritis Rheum. 2005;53(2):155-65.
3. Mills ES, Mathews WH. Interstitial pneumonitis in dermatomyositis. J Am Med Assoc. 1956;160(17):1467-70.
4. Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis. 2004;63(3):297-301.
5. Fathi M, Vikgren J, Boijsen M, Tylen U, Jorfeldt L, Tornling G, et al. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum. 2008;59(5):677-85.
6. Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28(4):859-90, viii.
7. Ghirardello A, Zampieri S, Iaccarino L, Tarricone E, Bendo R, Gambari PF, et al. Anti-Mi-2 antibodies. Autoimmunity. 2005;38(1):79-83.
8. Mahler M, Fritzler MJ. PM1-Alpha ELISA: the assay of choice for
the detection of anti-PM/Scl autoantibodies? Autoimmun Rev. 2009;8(5):373-8.
9. Hengstman GJ, van Engelen BG, van Venrooij W. Myositis specific autoantibodies: changing insights in pathophysiology and clinical associations. Curr Opin Rheumatol. 2004;16(6):692-9.
10. Marguerie C, Bunn CC, Beynon HL, Bernstein RM, Hughes JM, So AK, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med. 1990;77(282):1019-38.
11. Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70(6):360-74.
12. Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28(7):796-803.
13. Lega JC, Cottin V, Fabien N, Thivolet-Béjui F, Cordier JF. Interstitial lung disease associated with anti-PM/Scl or anti-aminoacyl-
tRNA synthetase autoantibodies: a similar condition? J Rheumatol. 2010;37(5):1000-9.
14. Mahler M, Raijmakers R, Dähnrich C, Blüthner M, Fritzler MJ. Clinical evaluation of autoantibodies to a novel PM/Scl peptide antigen. Arthritis Res Ther. 2005;7(3):R704-13.
15. Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. 1990;33(9):1361-70.
16. Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50(1):209-15.
17. Mathews MB, Reichlin M, Hughes GR, Bernstein RM. Anti-threonyl-tRNA synthetase, a second myositis-related autoantibody. J Exp Med. 1984;160(2):420-34.
18. Bunn CC, Bernstein RM, Mathews MB. Autoantibodies against alanyl-tRNA synthetase and tRNAAla coexist and are associated with myositis. J Exp Med. 1986;163(5):1281-91.
19. Targoff IN. Autoantibodies to aminoacyl-transfer RNA synthetases for isoleucine and glycine. Two additional synthetases are antigenic in myositis. J Immunol. 1990;144(5):1737-43.
20. Hirakata M, Suwa A, Nagai S, Kron MA, Trieu EP, Mimori T, et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol. 1999;162(4):2315-20.
21. Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford). 2007;46(6):1005-8.
22. Hashish L, Trieu EP, Sadanandan P, Targoff IN. Identification of autoantibodies to tyrosyl-tRNA synthetase in dermatomyositis with features consistent with anti-synthetase syndrome. Arthritis Rheum. 2005;52 (Suppl 9):s312.
23. Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum. 1980;23(8):881-8.
24. Marie I, Hachulla E, Chérin P, Dominique S, Hatron PY, Hellot MF, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum. 2002;47(6):614-22.
25. Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum. 2007;56(9):3125-31.
26. Hirakata M. Autoantibodies to aminoacyl-tRNA synthetases. Intern Med. 2005;44(6):527-8.
27. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-82.
28. Imbert-Masseau A, Hamidou M, Agard C, Grolleau JY, Chérin P. Antisynthetase syndrome. Joint Bone Spine. 2003;70(3):161-8.
29. Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity. 2005;38(1):73-8.
30. Mileti LM, Strek ME, Niewold TB, Curran JJ, Sweiss NJ. Clinical characteristics of patients with anti-Jo-1 antibodies: a single center experience. J Clin Rheumatol. 2009;15(5):254-5.
31. Mielnik P, Wiesik-Szewczyk E, Olesinska M, Chwalinska-Sadowska H, Zabek J. Clinical features and prognosis of patients with idiopathic inflammatory myopathies and anti-Jo-1 antibodies. Autoimmunity. 2006;39(3):243-7.
32. Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum.
1992;35(4):449-56.
33. Hirsch TJ, Enlow RW, Bias WB, Arnett FC. HLA-D related (DR)
antigens in various kinds of myositis. Hum Immunol. 1981;3(2):181-6.
34. Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry. 2000;68(4):472-8.
35. Koreeda Y, Higashimoto I, Yamamoto M, Takahashi M, Kaji K, Fujimoto M, et al. Clinical and pathological findings of interstitial lung disease patients with anti-aminoacyl-tRNA synthetase autoantibodies.
Intern Med. 2010;49(5):361-9.
36. Tillie-Leblond I, Wislez M, Valeyre D, Crestani B, Rabbat A, Israel-Biet D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax. 2008;63(1):53-9.
37. Schmidt WA, Wetzel W, Friedländer R, Lange R, Sörensen HF, Lichey HJ, et al. Clinical and serological aspects of patients with anti-Jo-1 antibodies--an evolving spectrum of disease manifestations. Clin Rheumatol. 2000;19(5):371-7.
38. Oddis CV, Medsger TA Jr, Cooperstein LA. A subluxing arthropathy associated with the anti-Jo-1 antibody in polymyositis/dermatomyositis. Arthritis Rheum. 1990;33(11):1640-5.
39. Schumacher HR, Schimmer B, Gordon GV, Bookspan MA, Brogadir S, Dorwart BB. Articular manifestations of polymyositis and dermatomyositis. Am J Med. 1979;67(2):287-92.
40. Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol. 2001;144(4):825-31.
41. Ponyi A, Constantin T, Garami M, András C, Tállai B, Váncsa A, et al. Cancer-associated myositis: clinical features and prognostic signs. Ann N Y Acad Sci. 2005;1051:64-71.
42. Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol. 2008;35(1):169-71.
43. Rozelle A, Trieu S, Chung L. Malignancy in the setting of the anti-synthetase syndrome. J Clin Rheumatol. 2008;14(5):285-8.
44. Asanuma Y, Koichihara R, Koyama S, Kawabata Y, Kobayashi S, Mimori T, et al. Antisynthetase syndrome associated with sarcoidosis. Intern Med. 2006;45(18):1065-8.
45. Diaco M, Ancarini F, Montalto M, Verrechia E, Evoli A, Servidei S, et al. Association of myasthenia gravis and antisynthetase syndrome: a case report. Int J Immunopathol Pharmacol. 2004;17(3):395-9.
46. Nouijai A, Ghazi M, Mounach A, Achemlal L, Bezza A, El Maghraoui A. Antisynthetase syndrome in a patient with ankylosing spondylitis. Joint Bone Spine. 2007;74(5):511-2.
47. Rovenský J, Kovalancík M, Payer J, Kohler K. Klinefelter syndrome with antisynthetase syndrome: why might they be associated? J Clin Rheumatol. 2003;9(1):62-3.
48. Szabo N, Lukacs S, Gunasekera W, Danko K. Rare association of antisynthetase syndrome and Kennedy's disease. Clin Rheumatol. 2008;27(10):1329-31.
49. Thickett DR, Millar AB. Drug-induced antisynthetase syndrome. Postgrad Med J. 1997;73(857):165-6.
50. Guglielmi S, Merz TM, Gugger M, Suter C, Nicod LP. Acute respiratory distress syndrome secondary to antisynthetase syndrome is reversible with tacrolimus. Eur Respir J. 2008;31(1):213-7.
51. Clawson K, Oddis CV. Adult respiratory distress syndrome in polymyositis patients with the anti-Jo-1 antibody. Arthritis Rheum. 1995;38(10):1519-23.
52. Fischer A, Swigris JJ, du Bois RM, Lynch DA, Downey GP, Cosgrove GP, et al. Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med. 2009;103(11):1719-24.
53. Sauty A, Rochat T, Schoch OD, Hamacher J, Kurt AM, Dayer JM, et al. Pulmonary fibrosis with predominant CD8 lymphocytic alveolitis and anti-Jo-1 antibodies. Eur Respir J. 1997;10(12):2907-12.
54. Yoshifuji H, Fujii T, Kobayashi S, Imura Y, Fujita Y, Kawabata D, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity. 2006;39(3):233-41.
55. Arnett FC, Hirsch TJ, Bias WB, Nishikai M, Reichlin M. The Jo-1 antibody system in myositis: relationships to clinical features and HLA. J Rheumatol. 1981;8(6):925-30.
56. Bernstein RM, Morgan SH, Chapman J, Bunn CC, Mathews MB, Turner-Warwick M, et al. Anti-Jo-1 antibody: a marker for myositis with interstitial lung disease. Br Med J (Clin Res Ed). 1984;289(6438):151-2.
57. Hochberg MC, Feldman D, Stevens MB, Arnett FC, Reichlin M. Antibody to Jo-1 in polymyositis/dermatomyositis: association with interstitial pulmonary disease. J Rheumatol. 1984;11(5):663-5.
58. Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum. 1996;39(2):292-6.
59. Walker EJ, Tymms KE, Webb J, Jeffrey P. Improved detection of anti-Jo-1 antibody, a marker for myositis, using purified histidyl-tRNA synthetase. J Immunol Methods. 1987;96(2):149-56.
60. Yoshida S, Akizuki M, Mimori T, Yamagata H, Inada S, Homma M. The precipitating antibody to an acidic nuclear protein antigen, the Jo-1, in connective tissue diseases. A marker for a subset of polymyositis with interstitial pulmonary fibrosis. Arthritis Rheum. 1983;26(5):604-11.
61. Watanabe K, Handa T, Tanizawa K, Hosono Y, Taguchi Y, Noma S, et al. Prevalence of autoantibodies to aminoacyl-transferRNA synthetases (ARS) among patients who were diagnosed with idiopathic interstitial pneumonias (IIPs) [abstract]. Am J Respir Crit Care Med. 2010;181:A2354.
62. Hervier B, Wallaert B, Hachulla E, Adoue D, Lauque D, Audrain
M, et al. Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatology (Oxford). 2010;49(5):972-6.
63. Kalluri M, Sahn SA, Oddis CV, Gharib SL, Christopher-Stine L, Danoff SK, et al. Clinical profile of anti-PL-12 autoantibody. Cohort study and review of the literature. Chest. 2009;135(6):1550-6.
64. Targoff IN, Arnett FC. Clinical manifestations in patients with antibody to PL-12 antigen (alanyl-tRNA synthetase). Am J Med. 1990;88(3):241-51.
65. Hirakata M, Nakamura Y, Okano Y, Suwa A, Inada S, Akizuki M,
et al. Anti-alanyl tRNA synthetase (PL-12) antibodies are associated with interstitial lung disease in Japanese patients [abstract]. Arthritis Rheum. 1995;38:S321.
66. Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum. 1996;26(1):459-67.
67. Handa T, Nagai S, Kawabata D, Nagao T, Takemura M, Kitaichi M, et al. Long-term clinical course of a patient with anti PL-12 antibody accompanied by interstitial pneumonia and severe pulmonary hypertension. Intern Med. 2005;44(4):319-25.
68. LaMedica G, Parodi A, Peris G, Rebora A. Polymyositis and pulmonary fibrosis associated with anti-PL-7 antibody. J Am Acad Dermatol. 1988;19(3):567-8.
69. Targoff IN, Arnett FC, Reichlin M. Antibody to threonyl-transfer RNA synthetase in myositis sera. Arthritis Rheum. 1988;31(4):515-24.
70. Sato S, Kuwana M, Hirakata M. Clinical characteristics of Japanese patients with anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibodies. Rheumatology (Oxford). 2007;46(5):842-5.
71. Okayasu K, Ohtani Y, Takemura T, Uchibori K, Tamaoka M, Furuiye M, et al. Nonspecific interstitial pneumonia (NSIP) associated with anti-KS antibody: differentiation from idiopathic NSIP. Intern Med. 2009;48(15):1301-6.
72. Frazier AR, Miller RD. Interstitial pneumonitis in association with polymyositis and dermatomyositis. Chest. 1974;65(4):403-7.
73. Salmeron G, Greenberg SD, Lidsky MD. Polymyositis and diffuse interstitial lung disease. A review of the pulmonary histopathologic findings. Arch Intern Med. 1981;141(8):1005-10.
74. Douglas WW, Tazelaar HD, Hartman TE, Hartman RP, Decker PA, Schroeder DR, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164(7):1182-5.
75. Nawata Y, Kurasawa K, Takabayashi K, Miike S, Watanabe N, Hiraguri M, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol. 1999;26(7):1527-33.
76. Schnabel A, Reuter M, Biederer J, Richter C, Gross WL. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32(5):273-84.
77. Tanaka F, Origuchi T, Migita K, Tominaga M, Kawakami A, Kawabe Y, et al. Successful combined therapy of cyclophosphamide and cyclosporine for acute exacerbated interstitial pneumonia associated with dermatomyositis. Intern Med. 2000;39(5):428-30.
78. Yoshida T, Koga H, Saitoh F, Sakamoto M, Harada M, Yoshida H, et al. Pulse intravenous cyclophosphamide treatment for steroid-resistant interstitial pneumonitis associated with polymyositis. Intern Med. 1999;38(9):733-8.
79. Hervier B, Masseau A, Mussini JM, Audrain M, Hamidou MA. Long-term efficacy of mycophenolate mofetil in a case of refractory antisynthetase syndrome. Joint Bone Spine. 2009;76(5):575-6.
80. López de la Osa A, Sánchez Tapia C, Arias Díaz M, Terrancle de Juan I. Antisynthetase syndrome with good response to mycophenolate mofetil [Article in Spanish]. Rev Clin Esp. 2007;207(5):269-70.
81. Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H,
Devulder B, et al. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol. 2001;28(10):2230-7.
82. Arsura EL, Greenberg AS. Adverse impact of interstitial pulmonary fibrosis on prognosis in polymyositis and dermatomyositis. Semin Arthritis Rheum. 1988;18(1):29-37.
83. Vandenbroucke E, Grutters JC, Altenburg J, Boersma WG, ter Borg EJ, van den Bosch JM. Rituximab in life threatening antisynthetase syndrome. Rheumatol Int. 2009;29(12):1499-502.
84. Ball EM, Savage EM, Pendleton A. Refractory anti-synthetase syndrome treated with rituximab. Rheumatology (Oxford). 2010;49(5):1013.
85. Sem M, Molberg O, Lund MB, Gran JT. Rituximab treatment of the anti-synthetase syndrome: a retrospective case series. Rheumatology (Oxford). 2009;48(8):968-71.
86. Suwa A, Hirakata M, Satoh S, Ezaki T, Mimori T, Inada S. A case of polymyositis with anti-OJ (isoleucyl-transfer RNA synthetase) antibodies. Clin Exp Rheumatol. 1999;17(6):755-6.
* Study carried out at the Interstitial Lung Disease Program and Autoimmune Lung Center, National Jewish Health, Denver, CO, USA.
Correspondence to: Joshua Solomon. Interstitial Lung Disease Program, National Jewish Health, 1400 Jackson Street, Denver, CO, 80206, USA.
Tel 1 303 398-1621. E-mail: solomonj@njhealth.org
Financial support: None.
Submitted: 21 September 2010. Accepted, after review: 21 October 2010.
** A versão completa em português deste artigo está disponível em www.jornaldepneumologia.com.br
About the authorsJoshua Solomon
M.D. Interstitial Lung Disease Program and Autoimmune Lung Center, National Jewish Health, Denver, CO, USA.
Jeffrey J. Swigris
M.D. Interstitial Lung Disease Program and Autoimmune Lung Center, National Jewish Health, Denver, CO, USA.
Kevin K. Brown
M.D. Interstitial Lung Disease Program and Autoimmune Lung Center, National Jewish Health, Denver, CO, USA.











 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket