
Continuous and bimonthly publication
ISSN (on-line): 1806-3756



Milena Baptistella Grotta, Elba Cristina de Sá Camargo Etchebere, Antonio Fernando Ribeiro, Juliana Romanato, Maria Ângela Gonçalves de Oliveira Ribeiro, José Dirceu Ribeiro
ABSTRACT
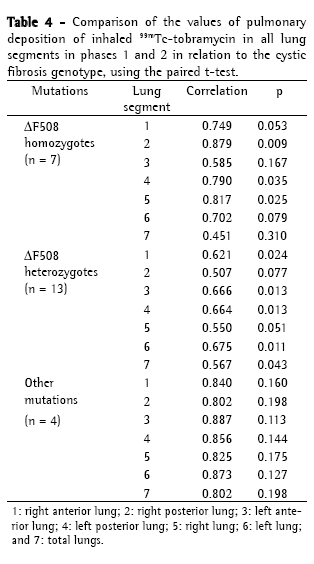
Objective: To evaluate whether respiratory therapy followed by the use of inhaled albuterol modifies the pulmonary deposition of inhaled tobramycin in patients with cystic fibrosis (CF) and whether pulmonary deposition correlates with disease severity or genotype. Methods: A prospective study was carried out including patients with CF older than 6 years of age and colonized with Pseudomonas aeruginosa. Exclusion criteria were pulmonary exacerbation, changes in therapy between the study phases and FEV1 < 25%. All patients were submitted to pulmonary scintigraphy by means of a scintillation camera equipped with a low-energy all-purpose collimator in order to evaluate drug penetration following the administration of inhaled 99mTc-tobramycin, as well as to pulmonary perfusion with 99mTc-macroaggregated albumin (phase 1). One month later, the same procedure was performed following respiratory therapy and administration of inhaled albuterol (phase 2). Results: We included 24 patients (12 males) aged 5-27 years (mean ± SD: 12.85 ± 6.64 years). The Shwachman score (SS) was excellent/good in 8 patients, moderate/fair in 16 and poor in 0. Genotyping revealed that 7 patients were ΔF508 homozygotes, 13 were ΔF508 heterozygotes; and 4 presented other mutations. In all patients, lung deposition of tobramycin decreased in phase 2, especially in those with moderate/fair SS (p = 0.017) and in heterozygotes (p = 0.043). Conclusions: The use of a respiratory therapy technique and the administration of inhaled albuterol immediately prior to the use of inhaled tobramycin decreased the pulmonary deposition of the latter in CF patients, and this reduction correlates with disease severity and genotype.
Keywords: Cystic fibrosis; Tobramycin; Respiratory therapy; Albuterol; Radionuclide imaging.
RESUMO
Objetivo: Avaliar se a fisioterapia respiratória seguida do uso de salbutamol inalatório modifica a deposição pulmonar de tobramicina inalatória em pacientes com fibrose cística (FC) e se a deposição pulmonar apresenta correlação com a gravidade da doença ou com o genótipo. Métodos: Um estudo prospectivo foi realizado com pacientes com FC maiores de 6 anos e colonizados por Pseudomonas aeruginosa. Os critérios de exclusão foram exacerbação pulmonar, mudança terapêutica entre as fases do estudo e FEV1 < 25%. Todos os pacientes foram submetidos à cintilografia pulmonar com câmara de cintilação com um colimador low energy all purpose para avaliar a penetração da droga após a inalação de tobramicina marcada com tecnécio (99mTc-tobramicina), e à perfusão pulmonar com 99mTc-macroagregados de albumina (fase 1). Após um mês, foi realizado o mesmo procedimento precedido de fisioterapia respiratória e administração de salbutamol inalatório (fase 2). Resultados: Foram incluídos 24 pacientes (12 masculinos) com idade variando de 5 a 27 anos (média ± dp: 12,85 ± 6,64 anos). O escore de Shwachman (ES) foi excelente/bom em 8 pacientes, moderado/regular em 8 e grave em 0. A genotipagem revelou que 7 pacientes eram ΔF508 homozigotos, 13 eram ΔF508 heterozigotos, e 4 apresentavam outras mutações. A deposição pulmonar da tobramicina foi menor na fase 2 em todos os pacientes, sendo menor nos pacientes com ES moderado/regular (p = 0,017) e também nos heterozigotos (p = 0,043). Conclusões: O uso de uma técnica de fisioterapia respiratória e a administração de salbutamol inalatório imediatamente antes do uso de tobramicina inalada diminuem a deposição pulmonar desta última em pacientes com FC, e esta redução tem correlação com a gravidade da doença e genótipo.
Palavras-chave: Fibrose cística; Tobramicina; Terapia respiratória; Albuterol; Cintilografia.
Introduction







Sociedade Brasileira de Pneumologia e Tisiologia
Secretaria do Jornal Brasileiro de Pneumologia
SCS Quadra 01, Bloco K, Salas 203/204 Ed. Denasa
Brasília - DF CEP: 70.398-900
Fone/fax: 08000 61 6218, (61) 3245 1030 e (61) 3245 6218
E-mail: jbp@sbpt.org.br
Development by: ![]()
© All rights reserved 2025 - Jornal Brasileiro de Pneumologia
 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket