ABSTRACT
Over the last five years, knowledge in the field of pulmonary hypertension has grown consistently and significantly. On the basis of various clinical studies showing the usefulness of new diagnostic tools, as well as the efficacy of new medications and drug combinations, new diagnostic and treatment algorithms have been developed. Likewise, in order to simplify the clinical management of patients, the classification of pulmonary hypertension has been changed in an attempt to group the various forms of pulmonary hypertension in which the diagnostic and therapeutic approaches are similar. The objective of this review was to discuss these modifications, based on the 2005 Brazilian guidelines for the management of pulmonary hypertension, emphasizing what has been added to the international guidelines.
Keywords:
Hypertension, pulmonary/diagnosis; Hypertension, pulmonary/therapy; Clinical protocols.
RESUMO
Ao longo dos últimos cinco anos, o conhecimento na área de hipertensão pulmonar evoluiu de forma consistente e significativa. Novos algoritmos diagnósticos e de tratamento foram desenvolvidos com base no resultado de diversos estudos clínicos que evidenciaram a utilidade de novas ferramentas, assim como a eficácia de novos medicamentos e de combinações. Da mesma forma, a classificação da hipertensão pulmonar evoluiu, na tentativa de agrupar as diferentes formas de hipertensão pulmonar que apresentam abordagens diagnósticas e terapêuticas semelhantes a fim de facilitar a condução clínica dos pacientes. Esta revisão visa discutir cada uma dessas modificações, tendo por base as diretrizes brasileiras para manejo da hipertensão pulmonar de 2005, ressaltando aquilo que foi acrescentado às diretrizes internacionais.
Palavras-chave:
Hipertensão pulmonar/diagnóstico; Hipertensão pulmonar/terapia; Protocolos clínicos.
IntroductionSince the latest pulmonary hypertension (PH) guidelines were published in this Journal in 2005, a new meeting of PH specialists was held in Dana Point, California, USA, in 2008, and various articles in the field PH have been published. Therefore, an update of certain aspects of PH diagnosis and treatment is necessary. In this article, we review what has changed in thediagnosis and treatment of PH in recent years.(1,2)
ScreeningPatients with complaints of dyspnea on exertion and chest pain, with or without dizziness, syncope, and signs of right heart failure of unknown cause, should be screened for PH. Various tests, which present a broad spectrum of sensitivity and specificity, can be used for the initial evaluation of patients suspected of having PH. Chest X-rays and electrocardiography (ECG), for instance, are tests that have low accuracy for the diagnosis of PH. Nevertheless, due to their broad availability and low cost, they can be employed in PH screening programs.
ECGIn patients with PH, ECG can show increased P wave amplitude (≥ 2.5 mm in the DII derivation), signs of right ventricular hypertrophy, right bundle branch block, right QRS axis deviation, and repolarization changes (right ventricular strain). Although a deviation greater than 100° has been shown to correlate well with hemodynamic measurements, its specificity for the diagnosis of PH has been shown to be low. Up to 13% of the patients with a diagnosis of PH confirmed by right heart catheterization (RHC) can initially present with normal ECG results (Figure 1).(3)
 Chest X-ray
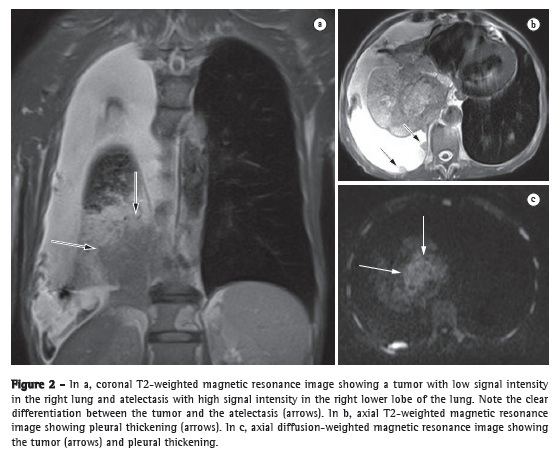
Chest X-rayA chest X-ray reveals hilar enlargement that reflects pulmonary artery (PA) dilation and cardiomegaly. Chest X-rays also play an important role in the diagnosis of other diseases, such as those that impair the lung parenchyma and can cause dyspnea (Figure 2a).
 Chest CT
Chest CTComputed angiotomography of the chest plays a significant role in the diagnostic evaluation of PH. The diameter of the PA trunk is significantly larger in patients with PH than in normal individuals and correlates well with PA pressure measurements.(4) Studies have shown that the diameter of the PA ranges from 32.6 mm to 33.2 mm in normal individuals. One group of authors found that a PA diameter > 33.2 mm has a 95% specificity for the diagnosis of PH (Figure 2b).(5)
EchocardiographyEchocardiography is the principal screening tool for PH. However, echocardiography is a test that has significant limitations, such as the fact that it is highly examiner-dependent and that a significant proportion of patients present with a poor acoustic window. Another limitation of echocardiography is that the estimation of PA systolic pressure (PASP) depends on the tricuspid regurgitant jet and right atrial pressure (RAP). In up to 10% of cases, it is impossible to measure the tricuspid regurgitant jet velocity and, consequently, to estimate PASP. Although some studies have shown a significant correlation between echocardiography findings and RHC values, one group of authors recently reported that the RAP and PASP values estimated by echocardiography differ significantly from those measured by RHC.(6) In that study, 65 patients referred for PH diagnosis or follow-up treatment underwent echocardiography and RHC one hour apart, meaning that the basal conditions of patients varied minimally. It was also shown that cardiac output (Qt) as measured by echocardiography is not useful and that echocardiography typically overestimates pressures. Therefore, PASP values as estimated by echocardiography should be used to screen for PH, rather than to diagnose it. In addition to PASP estimates, dilation and right ventricular dysfunction should be considered to constitute indirect signs of PH.
Despite its limitations, echocardiography continues to be the principal screening tool for PH because it is a noninvasive and readily available test, as well as being useful in identifying left heart malformations and diseases.
Because right ventricular function plays a significant role in the prognosis of patients with PH, it is necessary to measure right ventricular function appropriately. The characteristics of the right ventricle (RV) are quite different from those of the left ventricle (LV). Unlike the LV, which has thick, cone-shaped walls, the RV has thin, semilunar or crescent-shaped walls, and the myocardial mass of the RV is significantly lower and more trabecular than is that of the LV. The contraction pattern is also different; in the RV, longitudinal contraction of the myocardial fibers predominates, whereas, in the LV, spiral movement predominates. Therefore, it does not seem sufficient or appropriate to evaluate right ventricular function with the same tools used to evaluate left ventricular function.
New techniques for a better estimation of right ventricular function have been studied. The determination of tricuspid annular plane systolic excursion (TAPSE) has been shown to be a useful tool. This technique calculates the degree to which the pulmonary valve ring is shifted, in relation to the right ventricular apex during systole. A study comparing TAPSE and RHC measurements for the evaluation of right ventricular function showed that the measurements correlated well. The authors found that a TAPSE < 1.8 cm showed good accuracy in detecting right ventricular dysfunction and designated it a prognostic marker, because survival rates were lower in patients with a TAPSE < 1.8 cm than in those with a TAPSE ≥ 1.8 cm.(7)
Other techniques for the evaluation of right ventricular function, such as the comparison between the right ventricular area at systole and that at diastole-designated right ventricular fractional area change-and the comparison between the right ventricular end-diastolic area and the left ventricular end-diastolic area, have been studied and might prove useful in patients with PH.
Magnetic resonance imagingAdvances in the techniques for acquiring and processing magnetic resonance imaging of the heart have allowed three-dimensional evaluation of the RV and detailed tomographic visualization of its morphology. Cardiac magnetic resonance imaging (CMRI) creates a clear distinction between the myocardium and intracavitary blood, presenting well-defined myocardial and endocardial borders.(8) Because the RV presents the aforementioned particularities and CMRI allows a more detailed visualization of the RV, CMRI is currently considered the gold standard for a noninvasive evaluation of the RV.(9,10) Studies in which CMRI was used to evaluate patients with PH showed that, when compared with control group patients, PH patients presented with a significant increase in end-systolic and end-diastolic volumes, as well as in right ventricular muscle mass, together with a significant reduction in right ventricular ejection fraction. Other studies have shown ventricular septal bowing, together with a reduction in the LV volume in early diastole, revealing impaired left ventricular function associated with right ventricular dysfunction.(11) One group of authors demonstrated that the position of the septum, as determined by calculating its shift toward the LV, was accurate in predicting right ventricular systolic pressure.(12) Even without the use of contrast enhancement, CMRI allows excellent visualization of the PA, and it is possible to assess PA compliance and flow by means of the phase-contrast technique. In patients with PH, PA compliance values are significantly lower.(13) One study showed that measurements of pulsatility (which is related to compliance) can also correlate with the response to the NO test.(14) The measurement of PA velocity and the time it takes to reach the maximum velocity (acceleration time) are reduced in patients with PH, and these measurements are related to systolic volume as measured by RHC.(15) In addition, CMRI plays a role in the follow-up of patients with PH. Two studies used CMRI before treatment initiation and 6-12 months after treatment initiation. In one of the studies, the patients received epoprostenol, and in the other, they received bosentan.(16,17) In both studies, improvement in the six-minute walk test (6MWT) was significantly related to improvement in right ventricular function parameters, as determined by CMRI. In another study, CMRI was used before and after pulmonary thromboendarterectomy.(18) The study showed a significant reduction in myocardial mass, right ventricular end-systolic volume, and right ventricular end-diastolic volume, as well as increased left ventricular volumes, reflecting the reversion of ventricular remodeling and septal deviation, hemodynamic improvement having been achieved with the surgical procedure (Figure 2c).
Although CMRI is not widely available and its cost is still high, the role of CMRI in the diagnosis and follow-up of patients with PH is promising, because the test allows a better evaluation of right ventricular function, PA flow, and PA behavior.
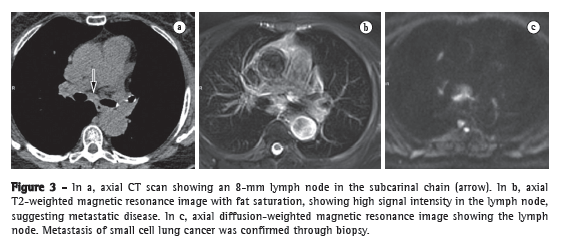
DiagnosisIf a patient suspected of having PH has been screened and signs consistent with increased pressure levels in the pulmonary circulation have been detected in the initial tests, the need to perform RHC to confirm the diagnosis of PH should be evaluated, because a definitive diagnosis of PH can only be established by invasive pressure measurements (Figure 3).
Since the last Brazilian consensus, there have been changes in the definition of PH. Before the meeting in Dana Point, CA, PH was defined as mean PA pressure (MPAP) ≥ 25 mmHg at rest or ≥ 30 mmHg during exercise, with pulmonary capillary pressure ≤ 15 mmHg. The group of specialists who reviewed the data that had been published up until the time of the meeting concluded that the data collected during exercise were extremely heterogeneous regarding the load used, the duration of the exercise, and the position of the patient during exercise, factors that might influence PA pressure measurements. Due to this lack of standardization, a decision was made to remove exercise-induced PH from the definition of PH. This does not mean that exercise-induced PH does not exist; it only means that the data collected to date are not sufficiently robust to provide a definition of exercise-induced PH values. This underscores the importance of conducting new studies in this field in order to provide an appropriate definition of exercise-induced PH.(2)
A review of 47 studies evaluating PA pressure in healthy volunteers showed that the MPAP at rest was 14.0 ± 3.3 mmHg. When individuals from different age brackets were compared, there was only a slight, less than significant, variation in the MPAP at rest.(19) Normal MPAP at rest was then defined as < 20 mmHg. The significance of a finding of pressure levels ranging from 20 mmHg to 25 mmHg remains unclear. Studies involving patients with COPD and pulmonary fibrosis showed that patients with MPAP > 17 mmHg had a worse prognosis than did those with MPAP < 17 mmHg and drew attention to the fact that MPAP < 25 mmHg might have clinical significance.(20) Values of MPAP at rest ≥ 25 mmHg are currently used to establish a diagnosis of PH.
Patients with MPAP ≥ 25 mmHg are diagnosed with PH, and, after the diagnosis has been established, it must be determined whether the PH is precapillary or postcapillary. If the PA occlusion pressure (PAOP) is ≤ 15 mmHg, the PH is classified as precapillary. If the PAOP is > 15 mmHg, the transpulmonary gradient (TPG) must be determined. The TPG is calculated be the difference between the MPAP and the PAOP. When this difference is ≤ 12 mmHg, the increase in the MPAP is considered passive, which means that the increase in the MPAP is caused exclusively by cardiac involvement. If the TPG is > 12 mmHg, the increase in the MPAP is disproportionate to the increase in left ventricular pressure, indicating that there is pulmonary vascular remodeling or another associated cause of increased MPAP (Chart 1 and Figure 3).

The acute test with a vasodilator should be performed during the initial hemodynamic evaluation in patients with precapillary PH. The test can be performed with NO, prostacyclin, or adenosine. The result is considered positive when there is a reduction in the MPAP of ≥ 10 mmHg and when values ≤ 40 mmHg are observed. A positive acute test result predicts the clinical and hemodynamic response to calcium channel blockers.(21,22)
After the presence of PH and its correct hemodynamic classification are confirmed by RHC, various tests should be performed in order to determine the specific etiology of PH. It should be highlighted that idiopathic PH is a differential diagnosis, and it is fundamental to follow an appropriate flowchart to facilitate the diagnostic investigation (Figure 4).
 Classification
ClassificationThere have been various changes in the clinical classification of PH. Although the basic structure of the 2003 Venice classification-five principal groups and their respective subgroups-was maintained, pathologies have been moved from one group or subgroup to another. In addition, some new groups or subgroups have been created, whereas others have been eliminated. These changes are described below. The former and current classifications are compared in Chart 2.(23)

The first group is still designated "pulmonary arterial hypertension" (PAH) and is divided into two subgroups: "idiopathic" and "heritable" (formerly "familial"). There have been no changes in subgroup 1.1, idiopathic PAH (IPAH), which still comprises the sporadic cases in which no risk factors for PAH are detected or in which there is no family history of PAH. Subgroup 1.2, heritable PAH, is subdivided as follows: 1.2.1-due to mutations in bone morphogenetic protein receptor, type 2 (BMPR2); 1.2.2-due to mutations in activin receptor-like kinase-1 or endoglin; and 1.2.3-of unknown cause. This new subdivision was necessary due to the importance of new genes associated with PH and of the description of mutations in the BMPR2 gene in 11-40% of the cases of IPAH; these cases, even without a family history of PAH, now characterize a subpopulation with hereditary disease, which makes the term "familial" inappropriate.
Subgroup 1.3 is now designated "drug- and toxin-induced" PAH. This change resulted from recent studies demonstrating the role of certain drugs in inducing PAH without changing its clinical course, as demonstrated for fenfluramine.(24) According to the new classification, subgroup 1.4 comprises conditions associated with the pathogenesis of PAH. Subgroup 1.4 subdivisions underwent small changes, HIV infection, portal hypertension, and persistent PH of the newborn remaining as subdivisions of this subgroup. The subgroup formerly known as "collagen vascular disease" is now designated "connective tissue diseases". The subgroup formerly known as "congenital systemic-pulmonary shunts" is now designated "congenital heart diseases". The subgroup designated "other" was eliminated from the current classification, and two new subgroups were created: subgroup 1.4.5, which now comprises patients with schistosomiasis; and subgroup 1.4.6, which comprises chronic hemolytic anemia, because the association between these pathologies and PAH has been shown to be important. Patients with schistosomiasis used to be allocated to the group of embolic diseases-group 4, in the previous classification-because it was believed that the mechanism that led to PH in this pathology was associated with the mechanic obstruction of pulmonary vessels by eggs of the parasite.
Anatomic pathology studies have demonstrated that the pulmonary involvement in schistosomiasis is similar to that found in IPAH, being accompanied by the development of plexiform lesions and hypertrophy of the tunica intima and tunica media, regardless of the obstruction by the parasite. Studies conducted in Brazil have also contributed to this change in the classification. One of these studies demonstrated that the clinical characteristics of patients with schistosomiasis and PH are similar to those of patients with IPAH.(25) The other study demonstrated that 7.7% of the patients with hepatosplenic schistosomiasis who were being treated at the University of São Paulo School of Medicine Hospital das Clínicas, located in the city of São Paulo, Brazil, had PH (4.6% of whom had precapillary PH).(26) Given the number of patients worldwide, schistosomiasis might become the leading cause of PH. Therefore, this change in the classification has a significant impact on Brazil and on all countries in which schistosomiasis is endemic. Concluding the changes in group 1, subgroup 1' (read "one prime") was created. This subgroup comprises pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis, because the histopathological features of these two entities have been shown to overlap. It is currently believed that they can represent different phases of the evolution of the same pathology. A decision was made to maintain the two entities in the group related to primarily arterial involvement, due to their degree of clinical response to the treatment with PAH-specific drugs, among other reasons.
Group 2 is designated "[PH] owing to left heart disease", in order to highlight the causal relationship between cardiac involvement and the development of PH, because this is potentially the most common cause of PH. This group was subdivided into three subgroups: 2.1-systolic dysfunction; 2.2-diastolic dysfunction; and 2.3-valvular disease.
In group 3, the term "associated with" was changed to "owing to", reinforcing the causal importance of pulmonary involvement. Therefore, group 3 is now designated "[PH] owing to lung disease and/or hypoxia", and its subgroups are as follows: COPD; interstitial lung disease; sleep-disordered breathing; alveolar hypoventilation disorders; chronic exposure to high altitude; developmental abnormalities; and a new subgroup, designated "other pulmonary diseases with mixed restrictive and obstructive pattern". The last subgroup comprises chronic bronchiectasis, cystic fibrosis, and a recently recognized syndrome in which fibrosis predominates in the lung bases and emphysema predominates in the lung apices. The prevalence of PH in patients with this syndrome is nearly
50%, and it is therefore necessary to emphasize this entity in the new classification.
Group 4, formerly known as "[PH] due to embolic disease, chronic thrombotic disease, or both", is now designated "chronic thromboembolic pulmonary hypertension" (CTEPH). The subdivision of obstruction into distal obstruction and proximal obstruction was removed from the classification, because the definitions of "proximal" and "distal" are difficult to standardize and vary among centers, making a definitive classification imprecise. This change should result in patients diagnosed with PH due to CTEPH being immediately referred to tertiary-care centers at which there are professionals with experience in performing thromboendarterectomy, so that operability can be determined by a multidisciplinary team.
Group 5 was changed from "miscellaneous" to "[PH] with unclear multifactorial mechanisms" and is now divided into four subgroups: "hematologic disorders"; "systemic disorders"; "metabolic disorders"; and a subgroup designated "others", which comprises a variety of conditions associated with PH.
TreatmentAfter the diagnosis of PH has been established by RHC and the disease has been clinically classified, PH treatment can be discussed, because the definition of the clinical group determines the treatment to be given. The general measures and the supportive therapy should be evaluated for any patient with PH; however, the largest amount of evidence, even regarding the general measures, is based on studies of patients with PAH, that is, group 1 patients.
General measures and supportive therapyAll patients diagnosed with PH should receive some general instructions. The patients should be instructed not to do heavy physical exercise and to limit physical activity when experiencing mild dyspnea. They should receive influenza vaccination and pneumococcal vaccination because infection is a major cause of morbidity and mortality in these patients. Female patients of childbearing age should be instructed to use contraceptive methods, because pregnancy significantly increases mortality in patients with PH. Although there is no consensus regarding the ideal type of contraception for patients with PH, it should be borne in mind that concomitant use of bosentan and oral contraceptives can reduce the effect of the latter. Patients in functional class III or IV, as well as those with hypoxemia (PaO2 < 60 mmHg), should use supplemental oxygen if flying or visiting areas at altitudes above 1,500-2,000 m.(27)
Supplemental oxygen therapy is indicated for patients with hypoxemia (PaO2 < 60 mmHg) and can be considered for patients who present symptomatic benefit from the correction of hypoxemia during physical exertion. The use of diuretics is indicated for all patients who present with signs of hypervolemia.(27)
The use of anticoagulants in patients with PH is controversial, because there have been no randomized controlled studies evaluating the effects of anticoagulation in these patients. The rationale for the indication of anticoagulation for these patients originates from the histopathological findings of microvascular thrombosis, activation of the coagulation system, and platelet dysfunction in patients with IPAH, which have led to the assumption that these patients present with a prothrombotic state. In a meta-analysis of the theme, conducted in 2006, the authors concluded that anticoagulation should be indicated, given that 5 of the 7 studies analyzed demonstrated that anticoagulation was beneficial. In the absence of any contraindications, the use of oral anticoagulants is indicated for patients with PH, with the objective of maintaining an international normalized ratio of 1.5-2.5. Attention should be given to patients with liver disease and scleroderma, because these patients might be at a higher risk for bleeding; likewise, attention should be given to the interaction between anticoagulants and the PAH-specific treatment. Some studies, for instance, have suggested that concomitant use of anticoagulants and sitaxsentan can increase the risk of bleeding.(28)
Group 1Most of the studies of PH treatment were conducted in patients with PAH; therefore, PH-specific treatment is, for the time being, restricted to group 1 patients. However, there has been no confirmation that all subgroups of patients with PAH respond to the specific drugs that are currently available. In general, the evidence discussed here is restricted to patients with IPAH, heritable PAH, drug-induced PAH, PAH associated with connective tissue diseases, or PAH associated with congenital heart diseases. There is also some evidence for patients with HIV infection. However, for patients with portopulmonary hypertension, schistosomiasis, or hemolytic anemia, it is still impossible to indicate the use of the same drugs, and clinical studies specifically designed to investigate those indications are needed.
If the patient presents pulmonary vasoreactivity, as assessed by the acute test, treatment with a calcium channel blocker should be initiated, and, if there is a sustained clinical response, the drug should be maintained, together with supportive treatment. The survival rates in patients who respond to the use of a calcium channel blocker are significantly higher than in those who do not respond well to the drug.(22) However, the use of calcium channel blockers in patients who do not present pulmonary vasoreactivity, as assessed by the acute test, can lead to a reduction in Qt and systemic vascular resistance without a reduction in the MPAP or pulmonary vascular resistance (PVR). Therefore, the use of a calcium channel blocker is contraindicated for patients who do not present pulmonary vasoreactivity or those who have not undergone the acute test, due to the risk of clinical deterioration. The calcium channel blocker to be used can be nifedipine, diltiazem, or amlodipine. However, in patients with high heart rates, diltiazem is the drug of choice. The treatment should be initiated with low doses, which should be progressively increased in accordance with the tolerance limit of the patient.
The classes of specific drugs that are approved for use in PAH patients, that is, in group 1 patients, are as follows: prostacyclin analogues; phosphodiesterase-5 inhibitors; and endothelin receptor antagonists.
Prostacyclin analoguesProstacyclin analogues constitute the first class of drugs to be approved for PH-specific treatment. Prostacyclin analogues can be administered intravenously, subcutaneously, orally, or by inhalation.
EpoprostenolIn an open randomized study conducted in 1996, clinical and hemodynamic improvements, as well as increased survival, were described in PAH patients who used epoprostenol in combination with conventional therapy (anticoagulation, diuretics, and oxygen therapy), when compared with those who used the conventional therapy in isolation.(29)
Although other studies have shown functional and hemodynamic improvements, the improvement in survival described in the aforementioned study has not been described elsewhere. Epoprostenol should be administered intravenously, through a tunneled catheter, and continuously, through a portable infusion pump, due to its short half-life. The most common side effects are jaw pain, flushing, diarrhea, nausea, and vomiting. Catheter-related complications, such as infection and thrombosis, as well as those related to the functioning of the equipment, have often been reported. Although epoprostenol is unavailable for use in Brazil, it is the only drug for functional class IV patients that has a grade of recommendation of A.
TreprostinilTreprostinil is a prostacyclin analogue whose half-life is longer than is that of epoprostenol, which allows treprostinil to be administered subcutaneously. In a randomized, placebo-controlled study, there was improvement in the symptoms, as well as slight but significant functional and hemodynamic improvements. Treprostinil has the advantage of not requiring an indwelling catheter, which avoids catheter-related complications. However, subcutaneous administration of treprostinil has been associated with pain at the injection site in 85% of the patients receiving the drug, and discontinuation of the drug is necessary in 8% of the cases. The speed at which the dose of the drug was increased was the principal causative factor for this side effect. Therefore, the dose should be increased slowly and progressively, and the site of injection should be changed every three days, in order to reduce this problem. The other side effects associated with epoprostenol can also occur in patients treated with treprostinil.(30) In one study, sustained hemodynamic and symptomatic improvements were observed during a mean follow-up period of 26 months. (27) Treprostinil is also currently unavailable for use in Brazil. The administration of the drug through inhalation and continuous intravenous administration is currently being evaluated.
IloprostIloprost is the prostacyclin analogue that is administered through inhalation. This route of administration has the advantage of acting on the pulmonary arteries that are in contact with ventilated regions; however, the drug must be inhaled 6-9 times a day and is commonly associated with the development of dry cough. Other side effects observed are the same as those of other prostacyclin analogues. A randomized, placebo-controlled study involving patients with PAH and CTEPH showed significant clinical improvement in a combined outcome that included physical exercise capacity, functional class in accordance with the New York Heart Association, and clinical deterioration for the group of patients treated with iloprost, who also presented hemodynamic stability during the study period.(31) Although it has been registered for use in Brazil, iloprost is not yet commercially available in the country.
BeraprostBeraprost is the only prostacyclin analogue that is available for oral administration. Although two studies have demonstrated an improvement in the six-minute walk distance (6MWD), this response was not sustained, and there was no hemodynamic response. The side effects of the drug are the same as those of other prostacyclin analogues.(28) In an open, uncontrolled study conducted in Japan, long-acting beraprost (a preparation designated TRK-100STP) was reported to produce clinical, functional, and hemodynamic improvement, its future use being therefore promising.(32) Beraprost is also currently unavailable for use in Brazil.
Phosphodiesterase-5 inhibitorsIncreased phosphodiesterase-5 in pulmonary arterioles and right ventricular myocytes has been demonstrated in patients with PH. The inhibition of this enzyme leads to an increase in the concentration of cyclic guanosine monophosphate, which promotes vasodilation, inhibits pulmonary artery remodeling due to its antiproliferative and pro-apoptotic effects, and seems to have a positive inotropic effect on the RV.(33)
Phosphodiesterase-5 inhibitors reduce PVR and lead to an increase in Qt, and the use of these drugs has been associated with clinical and functional improvement in patients with PH.(34,35) Two phosphodiesterase-5 inhibitors have been approved for use in patients with PH: sildenafil, approved in 2005; and tadalafil, approved in 2009. One study compared the use of increasing doses of sildenafil (20, 40, and 80 mg), administered three times a day, with the use of placebo. The study showed a significant but not dose-dependent increase in the 6MWD, as well as a significant, dose-dependent reduction in PVR.(36) The benefits of tadalafil have been demonstrated in another study, in which increasing doses of the drug were also compared with placebo. Only the 40-mg dose correlated with a significant increase in the 6MWD, an improvement in the markers of quality of life, and a slight increase in the time to clinical worsening.(37) Phosphodiesterase inhibitors are relatively safe and well tolerated. Tadalafil has the advantage of being administered only once daily. The major side effects of the drugs are headache, nasal congestion, dyspepsia, flushing, muscle pain, and epistaxis. Phosphodiesterase inhibitors are metabolized in the liver, and the use of protease inhibitors, such as ritonavir and saquinavir, can increase their bioavailability, and care should therefore be taken when prescribing this class of drugs to HIV-infected patients. Visual disorders, such as blurred vision, color changes, and photosensitivity, have been described, principally in patients with diabetic neuropathy or anterior ischemic optic neuropathy. Dilated eye examination is recommended before starting the treatment with this type of medication.(27)
Endothelin receptor antagonistsEndothelin-1 levels are elevated in the lung tissue and plasma of patients with PAH and scleroderma. Endothelin-1 acts by binding to endothelin receptors (ETA and ETB), promoting vasoconstriction and smooth muscle cell proliferation. Bosentan is a nonselective endothelin receptor antagonist, meaning that it blocks types A and B, and has been shown to be beneficial for patients with IPAH and for those with PAH associated with collagen disease, leading to an increase in exercise capacity and in the time to clinical worsening.(38) Other studies have reinforced the beneficial effects of bosentan, as well as showing hemodynamic and functional improvements.(27,39) One study showed that the use of bosentan in functional class lI patients is also beneficial.(40) The use of bosentan in these patients, who are less symptomatic, was shown to produce hemodynamic improvement and prevent clinical worsening.(40) The drug is generally well tolerated, and its principal side effect is an increase in hepatic enzyme levels, which requires that liver function be monitored throughout the treatment. The treatment should be initiated at a dose of 62.5 mg, twice daily, and, if the drug is well tolerated, the dose should be increased to 125 mg, twice daily. Monitoring through blood workup is also indicated, due to a report of anemia associated with the use of the drug.(27) Retrospective studies have also shown a reduction in the mortality associated with the use of endothelin receptor antagonists.
Another endothelin receptor antagonist is sitaxsentan, which is a specific inhibitor of the ETA receptor and has also been associated with better quality of life and functional capacity in patients with PH.(41) An open study comparing patients receiving sitaxsentan with those receiving bosentan demonstrated that the patients receiving sitaxsentan showed a trend toward reduced clinical worsening and better tolerance, with a lower increase in hepatic enzyme levels. A subgroup analysis has suggested that patients with PAH associated with connective tissue disease benefit significantly more from sitaxsentan than from bosentan.(42) The use of sitaxsentan significantly increases serum levels of dicumarol, requiring even closer monitoring of the coagulation profile during the treatment. This characteristic, together with case reports of complications related to the use of sitaxsentan, is what prevents this medication from being universally approved by the agencies that regulate the use of medications.
Yet another endothelin receptor antagonist is ambrisentan, which is also a selective ETA receptor antagonist and has been shown to produce a significant increase in the 6MWD and in the time to clinical worsening, as well as improving dyspnea and quality of life scores. It is of note that none of the patients who received treatment with ambrisentan presented with increased hepatic enzyme levels, and the drug also has the advantage of being administered only once daily. Another characteristic of the use of ambrisentan is the low interaction with other drugs, specifically with dicumarol and its derivatives, which allows safer concomitant use.(43)
However, it should be highlighted that there have been no controlled studies to determine which of the endothelin receptor antagonists, if any, is the most effective.
Combination therapyAlthough the therapeutic armamentarium for the treatment of PH has expanded greatly in recent years, a significant proportion of patients show no improvement or present clinical worsening during monotherapy. Until recently, there had been no studies confirming that the clinical response to combination therapy was effective or reporting that such therapy was well tolerated, although the concept of targeting different pathophysiological pathways was deemed logical in theory. Case reports and uncontrolled studies showed clinical improvement with and tolerance to the use of combination therapy. Consequently, randomized, placebo-controlled studies were conducted. Concomitant use of sildenafil in patients receiving epoprostenol was found to provide greater functional and hemodynamic improvement, as well as greater improvement in quality of life, together with an increase in the time to clinical worsening.(44)
Patients treated with the bosentan-epoprostenol combination showed a trend toward significant hemodynamic improvement, as well as good tolerance to the concomitant use of the two drugs. The lack of statistical significance is likely attributable to the small number of patients studied.
The only symptom that was more common in the group of patients treated with combination therapy was edema of the lower limbs, which was not attributed to right ventricular dysfunction, because the combination therapy produced a reduction in RAP. It is of note that there was a reduction in the side effects secondary to epoprostenol in the group of patients who also received bosentan, possibly due to the inhibition of the activation of the sympathetic system, which is characteristic of the use of epoprostenol, by bosentan.(45) Concomitant use of iloprost was well tolerated in patients receiving bosentan, who showed a trend toward an increase in the 6MWD, significantly improving functional parameters and increasing the time to clinical worsening.(46)
The use of combination therapy was therefore shown to be safe and effective. In cases in which the clinical response is inadequate or in which there is deterioration during monotherapy, concomitant use of another class of drugs should be indicated. Combination therapy can be initiated at the beginning of the treatment in cases in which the initial presentation is extremely severe. However, further studies are needed in order to confirm the true benefit of this approach.
Group 2The treatment of patients with PH caused by left heart disease should focus on compensating for the underlying heart disease, and the use of PH-specific treatment is therefore contraindicated for these patients.
Studies involving the use of bosentan and epoprostenol in this group of patients were terminated earlier than intended because the number of events observed in the groups of patients who received treatment was greater than was that observed in the placebo group. In addition, only one small-scale study has shown that the use of sildenafil is beneficial for the functional capacity of patients treated with the drug. These results therefore require corroboration from other studies, which might determine the true effectiveness and safety of the drugs that are currently available for this specific group of patients. Patients with a disproportionate increase in the MPAP, that is, those with TPG ≥ 12 mmHg, should be included in studies designed to that end.
Group 3Patients with parenchymal disease or hypoxemia should be primarily treated with oxygen therapy, and the treatment of the underlying disease should be optimized. The effectiveness of PH-specific drugs in this group of patients has yet to be confirmed, and the use of such drugs is currently contraindicated for these patients. When the MPAP is disproportionately high, that is, when parenchymal or functional involvement does not explain the degree of dyspnea and the MPAP at rest is higher than 40-45 mmHg, patients should be referred to a tertiary-care center, and the inclusion of such patients in studies is encouraged. However, PH-specific treatment should not be prescribed. It should be borne in mind that a finding of diastolic left ventricular dysfunction is not uncommon in these patients and might constitute another factor related to the pathogenesis of PH.
Group 4In all patients with PH, CTEPH should be ruled out. A ventilation/perfusion lung scintigraphy should always be performed, as seen in the diagnostic algorithm, and, if the result is normal, CTEPH can be ruled out. Angiotomography of the chest is useful for the evaluation of pulmonary circulation; however, it should not be used as an isolated tool for determining the operability of CTEPH. Patients with a diagnosis of CTEPH should receive anticoagulants and be referred to a tertiary-care center for the evaluation of the possibility of surgery. If the obstruction is operable, such patients should be referred for thromboendarterectomy. When a surgical intervention is contraindicated or cannot be performed, or when the patient presents with PH after thromboendarterectomy, the use of specific treatment seems beneficial.
A recent randomized controlled study demonstrated the hemodynamic benefit of the use of bosentan in patients with inoperable CTEPH, although the effect of the drug on functional capacity was not significant.(47) The use of sildenafil in patients with CTEPH for whom surgery is contraindicated has previously been described in a case series.(48) In addition, an open uncontrolled study conducted in 2007 and involving 104 patients showed good patient tolerance to the drug and a reduction in PVR, as well as improved function and 6MWD.(49) A small-scale but placebo-controlled study showed improved quality of life, as well as hemodynamic and functional improvement, in patients with residual PH after thromboendarterectomy or with distal CTEPH.(50) The decision to use specific medication in patients with CTEPH who cannot undergo surgery or who present with residual PH after surgical intervention should be made after an adequate evaluation of the case in a referral center, in order to rule out the hypothesis that these patients will benefit from the surgical approach. In addition, these patients should remain under close clinical monitoring.
Treatment initiation and follow-upAfter it has been decided whether patients should receive specific treatment, it is important to establish the clinical severity of the initial presentation of the disease. The fact that certain prognostic markers are major indicators of disease severity in patients with PH has been established in the literature. The following have been associated with a worse prognosis: functional classes III and IV; an increase in brain natriuretic peptide (BNP) or N-terminal-pro-BNP levels; 6MWD < 330 m; maximal oxygen uptake during the cardiopulmonary test < 12 mL/min/kg; and hemodynamic variables (RAP > 8 mmHg and cardiac index ≤ 2.0 L/min/m2 ).(27,51-53) Patients with markers of severity should be considered candidates for intravenous therapy in countries where this treatment is available. In localities where intravenous therapy is unavailable, combination therapy can be given at the initiation of treatment. However, it should be highlighted that there have been no clinical studies validating this approach; nevertheless, this approach has been considered in the latest international algorithms (Chart 3).

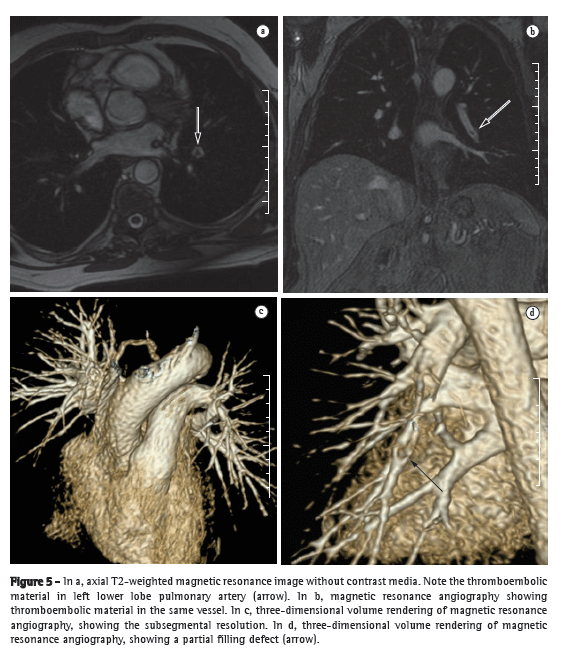
After the initiation of treatment, patients should be reevaluated, generally every 3-4 months, through analysis of the symptoms, physical examination findings, 6MWD, and BNP levels, in order to assess the response to treatment and decide the course of action. In localities where the 6MWT cannot be performed in a corridor, functional evaluation can be performed by means of a treadmill 6MWT, a protocol that has previously been validated for use in patients with PH.(54) If patients present clinical improvement or stabilization, as well as improvement or stabilization of the aforementioned markers, the treatment should be maintained; otherwise, an investigation should be performed in order to find the cause of treatment failure and avoid clinical deterioration. Infection, dietary noncompliance (excessive ingestion of salt or fluids), or the inappropriate use of the drugs are common causes of decompensation. If an evident cause is not found, a new hemodynamic evaluation can be performed, and, if hemodynamic worsening is confirmed, the specific treatment should be optimized by increasing the dose or adding another class of drugs. Patients who present with progressive worsening despite the optimized clinical treatment should be referred for an evaluation for lung transplantation (Figure 5).(55)
 Future perspectives
Future perspectivesThere have been significant advances in recent years, and existing concepts, the levels of evidence of which had not been sufficient to allow further extrapolations, have been consolidated, making it possible to construct new diagnostic and therapeutic algorithms. The prospect of effective therapeutic alternatives for the various PH groups is excellent, and new pathophysiological pathways with therapeutic potential have been discovered, providing the spark for the development of new classes of drugs that might be added to the existing therapeutic armamentarium.
This development has always been based on research, of increasing quality, which generates increasingly robust evidence. In the coming years, the expectation is that this characteristic will be increasingly present in the field of PH.(56)
References1. Sociedade Brasileira de Pneumologia e Tisiologia. Classificação e avaliação diagnóstica da hipertensão pulmonar. J Bras Pneumol. 2005;31(2 Suppl):S1-S8.
2. Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S55-66.
3. Ahearn GS, Tapson VF, Rebeiz A, Greenfield JC Jr. Electrocardiography to define clinical status in primary pulmonary hypertension and pulmonary arterial hypertension secondary to collagen vascular disease. Chest. 2002;122(2):524-7.
4. Karazincir S, Balci A, Seyfeli E, Akoğlu S, Babayiğit C, Akgül F, et al. CT assessment of main pulmonary artery diameter. Diagn Interv Radiol. 2008;14(2):72-4.
5. Edwards PD, Bull RK, Coulden R. CT measurement of main pulmonary artery diameter. Br J Radiol. 1998;71(850):1018-20.
6. Fisher MR, Forfia PR, Chamera E, Housten-Harris T, Champion HC, Girgis RE, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179(7):615-21.
7. Forfia PR, Fisher MR, Mathai SC, Housten-Harris T, Hemnes AR, Borlaug BA, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174(9):1034-41.
8. Steendijk P. Right ventricular function and failure: methods, models, and mechanisms. Crit Care Med. 2004;32(4):1087-9.
9. Woods J, Monteiro P, Rhodes A. Right ventricular dysfunction. Curr Opin Crit Care. 2007;13(5):532-40.
10. Kosiborod M, Wackers FJ. Assessment of right ventricular morphology and function. Semin Respir Crit Care Med. 2003;24(3):245-62.
11. Marcus JT, Vonk Noordegraaf A, Roeleveld RJ, Postmus PE, Heethaar RM, Van Rossum AC, et al. Impaired left ventricular filling due to right ventricular pressure overload in primary pulmonary hypertension: noninvasive monitoring using MRI. Chest. 2001;119(6):1761-5.
12. Dellegrottaglie S, Sanz J, Poon M, Viles-Gonzalez JF, Sulica R, Goyenechea M, et al. Pulmonary hypertension: accuracy of detection with left ventricular septal-to-free wall curvature ratio measured at cardiac MR. Radiology. 2007;243(1):63-9.
13. Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, et al. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest. 2007;132(6):1906-12.
14. Jardim C, Rochitte CE, Humbert M, Rubenfeld G, Jasinowodolinski D, Carvalho CR, et al. Pulmonary artery distensibility in pulmonary arterial hypertension: an MRI pilot study. Eur Respir J. 2007;29(3):476-81.
15. Sanz J, Kuschnir P, Rius T, Salguero R, Sulica R, Einstein A, et al. Pulmonary arterial hypertension: noninvasive detection with phase-contrast MR imaging. Radiology. 2007;243(1):70-9.
16. Chin KM, Kingman M, de Lemos JA, Warner JJ, Reimold S, Peshock R, et al. Changes in right ventricular structure and function assessed using cardiac magnetic resonance imaging in bosentan-treated patients with pulmonary arterial hypertension. Am J Cardiol. 2008;101(11):1669 72.
17. Roeleveld RJ, Vonk-Noordegraaf A, Marcus JT, Bronzwaer JG, Marques KM, Postmus PE, et al. Effects of epoprostenol on right ventricular hypertrophy and dilatation in pulmonary hypertension. Chest. 2004;125(2):572-9.
18. Reesink HJ, Marcus JT, Tulevski II, Jamieson S, Kloek JJ, Vonk Noordegraaf A, et al. Reverse right ventricular remodeling after pulmonary endarterectomy in patients with chronic thromboembolic pulmonary hypertension: utility of magnetic resonance imaging to demonstrate restoration of the right ventricle. J Thorac Cardiovasc Surg. 2007;133(1):58-64.
19. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888-94.
20. Kovacs G, Maier R, Aberer E, Brodmann M, Scheidl S, Tröster N, et al. Borderline pulmonary arterial pressure is associated with decreased exercise capacity in scleroderma. Am J Respir Crit Care Med. 2009;180(9):881-6.
21. Sitbon O, Humbert M, Jagot JL, Taravella O, Fartoukh M, Parent F, et al. Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur Respir J. 1998;12(2):265-70.
22. Sitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105-11.
23. Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43-54.
24. Souza R, Humbert M, Sztrymf B, Jaïs X, Yaïci A, Le Pavec J, et al. Pulmonary arterial hypertension associated with fenfluramine exposure: report of 109 cases. Eur Respir J. 2008;31(2):343-8.
25. Lapa MS, Ferreira EV, Jardim C, Martins Bdo C, Arakaki JS, Souza R. Clinical characteristics of pulmonary hypertension patients in two reference centers in the city of Sao Paulo [Article in Portuguese]. Rev Assoc Med Bras. 2006;52(3):139-43.
26. Lapa M, Dias B, Jardim C, Fernandes CJ, Dourado PM, Figueiredo M, et al. Cardiopulmonary manifestations of hepatosplenic schistosomiasis. Circulation. 2009;119(11):1518-23.
27. Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC); European Respiratory Society (ERS); International Society of Heart and Lung Transplantation (ISHLT); Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34(6):1219-63.
28. Johnson SR, Mehta S, Granton JT. Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur Respir J. 2006;28(5):999-1004.
29. Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;334(5):296-302.
30. Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800-4.
31. Olschewski H, Simonneau G, Galiè N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322-9.
32. Kunieda T, Nakanishi N, Matsubara H, Ohe T, Okano Y, Kondo H, et al. Effects of long-acting beraprost sodium (TRK-100STP) in Japanese patients with pulmonary arterial hypertension. Int Heart J. 2009;50(4):513-29.
33. Archer SL, Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009;361(19):1864-71.
34. Fernandes CJ, Jardim C, Carvalho LA, Farias AQ, Filho MT, Souza R. Clinical response to sildenafil in pulmonary hypertension associated with Gaucher disease. J Inherit Metab Dis. 2005;28(4):603-5.
35. de Carvalho AC, Hovnanian AL, Fernandes CJ, Lapa M, Jardim C, Souza R. Tadalafil as treatment for idiopathic pulmonary arterial hypertension. Arq Bras Cardiol. 2006;87(5):e195-7.
36. Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148-57. Erratum in: N Engl J Med. 2006;354(22):2400-1.
37. Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894-903.
38. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896-903. Erratum in: N Engl J Med. 2002;346(16):1258.
39. Souza R, Jardim C, Martins B, Cortopassi F, Yaksic M, Rabelo R, et al. Effect of bosentan treatment on surrogate markers in pulmonary arterial hypertension. Curr Med Res Opin. 2005;21(6):907-11.
40. Galiè N, Rubin Lj, Hoeper M, Jansa P, Al-Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371(9630):2093-100.
41. Souza R, Martins BC, Jardim C, Cortopassi F, Fernandes CJ, Pulido T, et al. Effect of sitaxsentan treatment on quality of life in pulmonary arterial hypertension. Int J Clin Pract. 2007;61(1):153-6.
42. Benza RL, Barst RJ, Galie N, Frost A, Girgis RE, Highland KB, et al. Sitaxsentan for the treatment of pulmonary arterial hypertension: a 1-year, prospective, open-label observation of outcome and survival. Chest. 2008;134(4):775-82.
43. Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010-9.
44. Simonneau G, Rubin LJ, Galiè N, Barst RJ, Fleming TR, Frost AE, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 200;149(8):521-30.
45. Humbert M, Barst RJ, Robbins IM, Channick RN, Galiè N, Boonstra A, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24(3):353-9.
46. McLaughlin VV, Oudiz RJ, Frost A, Tapson VF, Murali S, Channick RN, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174(11):1257-63.
47. Jaïs X, D'Armini AM, Jansa P, Torbicki A, Delcroix M, Ghofrani HA, et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol. 2008;52(25):2127-34.
48. Reichenberger F, Voswinckel R, Enke B, Rutsch M, El Fechtali E, Schmehl T, et al. Long-term treatment with sildenafil in chronic thromboembolic pulmonary hypertension. Eur Respir J. 2007;30(5):922-7.
49. Dias BA, Jardim C, Hovnanian A, Fernandes CJ, Souza R. Chronic thromboembolic pulmonary hypertension: diagnostic limitations. J Bras Pneumol. 2008;34(7):532-6.
50. Suntharalingam J, Treacy CM, Doughty NJ, Goldsmith K, Soon E, Toshner MR, et al. Long-term use of sildenafil in inoperable chronic thromboembolic pulmonary hypertension. Chest. 2008;134(2):229-36.
51. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114(13):1417-31.
52. Souza R, Bogossian HB, Humbert M, Jardim C, Rabelo R, Amato MB, et al. N-terminal-pro-brain natriuretic peptide as a haemodynamic marker in idiopathic pulmonary arterial hypertension. Eur Respir J. 2005;25(3):509-13.
53. Souza R, Jardim C, Carvalho C, Rubenfeld G. The role of NT-proBNP as a prognostic marker in pulmonary hypertension. Chest. 2006;130(5):1627; author reply 1627-8.
54. Camargo VM, Martins Bdo C, Jardim C, Fernandes CJ, Hovnanian A, Souza R. Validation of a treadmill six-minute walk test protocol for the evaluation of patients with pulmonary arterial hypertension. J Bras Pneumol. 2009;35(5):423-30.
55. Barst RJ, Gibbs JS, Ghofrani HA, Hoeper MM, McLaughlin VV, Rubin LJ, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S78-84.
56. Souza R, Jardim C. Trends in pulmonary arterial hypertension. Eur Resp Rev 2009;18(111):7-12.
* Study carried out at the Pulmonary Circulation Unit, Department of Pulmonology, Instituto do Coração, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo - InCor/HC-FMUSP, Heart Institute/University of São Paulo School of Medicine Hospital das Clínicas - São Paulo, Brazil.
Correspondence to: Rogério de Souza. Avenida Dr. Enéas de Carvalho Aguiar, 44, 5º andar, Bloco II, CEP 05403-000, São Paulo, SP, Brasil.
Tel/Fax: 55 11 3069-5695. E-mail: rogerio.souza@incor.usp.br
Financial support: None.
Submitted: 6 March 2010. Accepted, after review: 6 July 2010.
About the authorsSusana Hoette
Pulmonologist. Pulmonary Hypertension Group, Department of Pulmonology, Instituto do Coração, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo - InCor/HC-FMUSP, Heart Institute/University of São Paulo School of Medicine Hospital das Clínicas - São Paulo, Brazil.
Carlos Jardim
Attending Physician. Pulmonary Hypertension Group, Department of Pulmonology, Instituto do Coração, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo - InCor/HC-FMUSP, Heart Institute/University of São Paulo School of Medicine Hospital das Clínicas -São Paulo, Brazil.
Rogério de Souza
Tenured Professor. Department of Pulmonology, Instituto do Coração, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo - InCor/HC-FMUSP, Heart Institute/University of São Paulo School of Medicine Hospital das Clínicas - São Paulo, Brazil.






 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket




