ABSTRACT
Objective: To study correlations among pulmonary function, chest radiology and clinical status in cystic fibrosis. Methods: A retrospective
cross-sectional study was performed to evaluate chest X-rays and clinical charts of patients treated at the Hospital de Clínicas de Porto
Alegre. Spirometry findings, Shwachman-Kulczycki (S-K) scores and Brasfield scores were analyzed. Results: The final sample consisted
of 40 patients (mean age 9.72 ± 3.27). The following mean S-K scores were obtained: total, 80.87 ± 10.24; general activity, 24.75 ± 1.1;
physical examination, 18.87 ± 4.59; nutrition, 21.87 ± 4.18; radiology, 15.37 ± 5.23. The mean Brasfield score was 18.2 ± 4. The pulmonary
function test results, in percentage of predicted, were as follows: forced vital capacity (FVC), 82.99 ± 14.36%; forced expiratory volume
in one second (FEV1), 83.62 ± 18.26%; and forced expiratory flow between 25 and 75% of FVC (FEF25-75), 74.63 ± 2.53%. The S-K score
correlated moderately with FVC, whereas it correlated strongly with FEV1 and FEF25-75. The Brasfield score correlated strongly with the S-K
total and radiology score, whereas it correlated moderately with pulmonary function. Physical examination correlated moderately with FVC,
FEV1 and FEF25-75; as did nutrition with FEF25-75; and radiology with FEV1 and FEF25-75. General activity was the domain that had the greatest
influence on the total S-K score. Conclusions: These two scoring systems are complementary, correlating with each other, as well as with
pulmonary function tests. The radiology domain of the S-K scoring system is a good alternative to the Brasfield score.
Keywords:
Cystic fibrosis; Spirometry; Lung/radiography.
RESUMO
Objetivo: Estudar as relações entre função pulmonar, radiologia de tórax e estado clínico em fibrose cística. Métodos: Em estudo transversal
e retrospectivo, analisaram-se prontuários e radiografias de pacientes do Hospital de Clínicas de Porto Alegre. Foram estudados as
espirometrias e os escores de Shwachman-Kulczycki (S-K) e de Brasfield. Resultados: A amostra final constituiu-se de 40 pacientes com
média de idade de 9,72 ± 3,27. Foram obtidas as seguintes médias dos escores de S-K: total, 80,87 ± 10,24; atividade geral, 24,75 ± 1,1;
exame físico, 18,87 ± 4,59; nutrição, 21,87 ± 4,18; e radiologia, 15,37 ± 5,23. A média do escore de Brasfield foi de 18,2 ± 4. As médias das
variáveis espirométricas foram, em porcentagem do previsto: capacidade vital forçada (CVF), 82,99 ± 14,36%; volume expiratório forçado
no primeiro segundo (VEF1), 83,62 ± 18,26%; e fluxo expiratório forçado entre 25 e 75% da CVF (FEF25-75), 74,63 ± 2,53%. O escore de S-K
correlacionou-se moderadamente com a CVF e fortemente com VEF1 e FEF25-75. O escore de Brasfield correlacionou-se fortemente com o
escore de S-K total e da radiologia, e moderadamente com a função pulmonar. O escore do exame físico correlacionou-se moderadamente
com CVF, VEF1 e FEF25-75, bem como o da nutrição com FEF25-75 e o da radiologia com VEF1 e FEF25-75. A atividade geral foi a categoria que
mais contribuiu para a pontuação total do S-K. Conclusões: Esses escores utilizados para a monitorização da fibrose cística são complementares,
correlacionando-se entre si e com as provas funcionais. A categoria radiologia do escore de S-K é um bom substituto para o escore
de Brasfield.
Palavras-chave:
Fibrose cística; Espirometria; Pulmão/radiografia.
IntroductionCystic fibrosis is the most common of the lethal genetic diseases in the Caucasian population, and the reported incidence is 1:2500 births.(1) Its gene encodes a 1480-amino acid protein known as the cystic fibrosis transmembrane regulator (CFTR) protein, located in the submucosal glands of the tracheobronchial tree, the pancreatic ducts, the hepatic ducts, the intestinal crypts, the renal tubes, the genitourinary system and the sweat glands.(2) The CFTR protein has multiple functions, which are only partially known. A mutation in the gene results in a modification in the structure of the protein, and over 1500 different mutations(3) have been determined to date. Dysfunction of the CFTR protein leads to a disturbance in chloride transport through the epithelium, resulting in dehydration of the mucosal surfaces with the formation of dense mucus which favors the advent of infections, resulting in progressive loss of pulmonary function. Pulmonary involvement is responsible for over two thirds of all cystic fibrosis-related deaths.(4)
Radiological features such as hyperinflation, bronchial thickening, mucoid impaction, bronchiectasis and atelectasis appear early.(5) Function tests are characterized by an increase in total lung capacity, functional residual capacity and residual volume, in addition to early involvement of the distal flows and the presence of bronchial hyperreactivity.(6) The principal confirmation test is the sweat test, and two tests with chloride tests are necessary, each with >60 mEq/L (for children) or >80 mEq/L (for adults).(7) In genetic analysis, the identification of two gene mutations confirms the diagnosis, but its absence does not rule it out. Treatment is based on the use of antibiotics, fluidification of pulmonary secretion, respiratory therapy, pancreatic supplementation and maintaining adequate nutritional status, thus increasing the survival of these patients considerably.(4)
Disease progression is assessed through pulmonary function tests of forced vital capacity (FVC), forced expiratory volume in one second (FEV1) and forced expiratory flow between 25 and 75% of FVC (FEF25-75), the last presenting the earliest alterations,(8,9) as well as through imaging and collection of clinical data. Chest X-ray has long been the first diagnostic imaging method used in the evaluation of pulmonary involvement and is adequate for the detection of the most significant lesions.(10,11)
The severity of the respiratory disease is the most important isolated factor, affecting the survival of these patients. Since it is impossible to efficiently monitor the disease based on pulmonary function tests, imaging tests and clinical data simultaneously, it is necessary to analyze the correlations among these three methods so that the physician can give each of them the appropriate weight. Numerous scoring systems have been used to more objectively classify the severity of the pulmonary disease.(11-15) The Shwachman-Kulczycki clinical score(16) and the Brasfield radiological score(17) are widely used. There are numerous reports in the literature of correlations between these scores and pulmonary function.(18-24)
The objective of this study was to correlate functional data, radiological alterations and the clinical status of cystic fibrosis patients.
MethodsWe analyzed the clinical charts and radiological documents of 40 cystic fibrosis patients aged 5-16 years out of a total of 98 patients under follow-up treatment at the Cystic Fibrosis Center of the Porto Alegre Hospital de Clínicas between January of 2000 and December of 2003. The diagnosis of cystic fibrosis was made in accordance with the consensus criteria.(7) Pediatric patients with stable disease (without exacerbation) who, in their most recent annual physical examination, had been submitted to a conventional chest X-ray, spirometry (including a pharmacodynamic test) and application of the Shwachman-Kulczycki score (Table 1) were included in the study. Exclusion criteria were as follows: having bronchial asthma; having allergic bronchopulmonary aspergillosis; currently being a smoker; and having previously undergone transplantation or pulmonary resection.
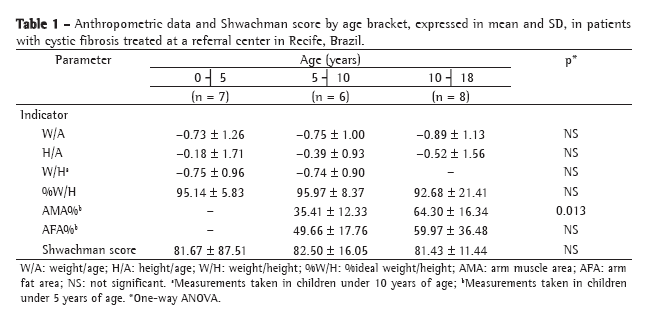
This was a cross-sectional retrospective study. It was calculated that a sample size of 38 patients was needed in order to estimate correlations equal to or greater than 0.50 with a level of significance of α = 0.05 and a statistical power (1-ß) of 90%. Anteroposterior and lateral chest X-rays were analyzed according to the Brasfield score (Chart 1), in a blinded fashion, by a pediatric radiologist of renowned experience. The Brasfield score consists of five domains representing the characteristic radiological aspects of cystic fibrosis: air trapping; linear markings (bronchial wall thickening); nodular cystic lesions (bronchiectasis); large lesions (atelectasis and pneumonia); and general severity. The first three radiological alterations are scored according to severity from 0 to 4; the last two, from 0 to 5. The final score was obtained by totaling the five subscores and then subtracting the result from 25 (3 is the most severe score possible).

Spirometry (including a pharmacodynamic test) was performed by one single technician with experience in pediatric tests and as per the standards recommended by the First Brazilian Consensus on Spirometry.(8) The tests analyzed were FVC, FEV1 and FEF25-75, all expressed as the percentage of the predicted value based on gender, age and height in accordance with the equations devised by Zapletal.(25) A Master Screen spirometer (Jaeger, Würzburg, Germany) was used, according to the routine of the Pulmonary Physiology Unit (Pulmonary Function Laboratory) of the Porto Alegre Hospital de Clínicas Pulmonology Department.
The Shwachman-Kulczycki score is divided into four domains, each with five possible scores, according to the degree of impairment: general activity; physical examination; nutrition; and radiological findings. The four scores are totaled in order to obtain the final score, by which the condition of the patient is categorized as excellent (86-100), good (71-85), average (56-70), poor (41-55) or severe (≤40). Scoring was performed by the physician responsible for the treatment of the patient at their annual physical examination, and each of the four pediatric pulmonologists of the team monitored each patient throughout the course of the disease.
Brasfield and Shwachman-Kulczycki scores, function test results and patient ages are expressed as means and standard deviations. The five domains of the Brasfield score are expressed as medians and interquartile ranges. Pearson's correlation coefficient was used in order to compare pulmonary function tests with scores. Multiple linear regression analysis was performed in order to determine which variables most influenced the Shwachman-Kulczycki total score. The level (alpha) of statistical significance was set at 0.05, and the statistical power (1-ß) was set at 90%. A database was created using the Excel program, and the software Statistical Package for the Social Sciences for Windows, version 12.0 (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis of the data. The protocol of the present study was approved by the Science and Ethics Committee of the Porto Alegre Hospital de Clínicas.
ResultsPatients who were too young to be submitted to spirometry (under 5 years of age) and patients for whom a complete evaluation through simultaneous performance of tests could not be performed were excluded, as were those who presented exacerbation of the disease. The sample studied comprised 40 patients (19 boys and 21 girls) aged 5-16 years. Regarding the Shwachman-Kulczycki total score, 62.5% of the 40 patients presented scores equal to or greater than 80, with a mean score of 80.9 ± 10.2. In the general activity domain, 95% presented a score of 25, the lowest score being 20 (mean, 24.7 ± 1.1). In the physical examination domain, 40% of the children presented a score of 15, and 55% presented scores ≥20 (mean, 18.9 ± 4.6). In the nutrition domain, 32.5% presented a score of 20, and 55% presented a score of 25 (mean, 21.9 ± 4.2). In the radiology domain, 40% presented a score of 10, and 10% presented a score of 25 (mean, 15.4 ± 5.2). This group of patients presented very good pulmonary function, with a mean FEV1 of 83.6 ± 18.2% and a mean FEF25-75 of 74.6 ± 42.5%. Regarding the Brasfield score, the mean was also high (18.2 ± 4, bearing in mind that the maximum score is 25). Data for the variables studied can be seen in Table 2.
The Shwachman-Kulczycki score was found to correlate positively with FVC (r = 0.35), as well as (more strongly) with FEV1 (r = 0.50) and FEF25-75 (r = 0.54). The Brasfield radiological score correlated strongly and positively with the Shwachman-Kulczycki score (r = 0.62) and with the radiology domain of the same (r = 0.64). However, when compared with functional parameters, the Brasfield score correlated only moderately with FVC (r = 0.32), FEV1 (r = 0.36) and FEF25-75 (r = 0.35). Correlation coefficients are shown in Table 3 and Figure 1.

When pulmonary function was correlated with the domains of the Shwachman-Kulczycki score, positive, moderate correlations were found between the physical examination domain and FVC (r = 0.34 p = 0.03), FEV1 (r = 0.39 p = 0.01) and FEF25-75 (r = 0.42 p < 0.01); between the nutrition domain and FEF25-75 (r = 0.32 p = 0.04); and between the radiology domain and both FEV1 (r = 0.42 p < 0.01) and FEF25-75 (r = 0.38 p = 0.01). The p was not significant when the general activity domain was correlated with pulmonary function, when the nutrition domain was correlated with both FVC and FEV1 and when the radiology domain was correlated with FVC.
Regarding the contribution of each Shwachman-Kulczycki score domain to the total score, the physical examination domain correlated significantly with the general activity domain (r = 0.32; p = 0.04) and with the radiology domain (r = 0.50; p < 0.01), and was therefore excluded from the analysis. The general activity domain was the one that contributed the most to the total score, each point increasing the final score by 1.93 (p = 0.02).
DiscussionCystic fibrosis is a complex disease, and scoring systems are necessary in order to objectively assess its severity,(14,26,27) since most clinical aspects evaluated can be affected by the subjectivity of the examiner. The ideal score would classify patients and clearly predict their clinical course in a simple manner, adapting itself to the routine treatment.
The most widely used score is that created by Shwachman-Kulczycki.(28) Although it is easily applied by the attending physician, most of its domains are based on subjective information. Created based on a study in which 105 patients were monitored for 5 years, which exposed the need for a system to evaluate the severity of this disease,(16) the Shwachman-Kulczycki score provides a perception of the overall clinical status of the patient. This score presents high intra- and inter-rater reliability.(11) However, it lacks a domain that evaluates pulmonary function.
In the present study, the total Shwachman-Kulczycki score correlated positively with the scores for each of its four domains, and physical activity was the aspect that most influenced total score. This corroborates our idea that the initial alterations are in the morphological aspect of the disease, as seen on imaging tests. Later, there are alterations in pulmonary function, although the patient is still capable of engaging in physical activity. Finally, the capacity for physical exertion is impaired. Despite the inevitable loss of pulmonary function, patients with the mild or moderate form of the disease are still capable of engaging in physical activity, since factors other than FEV1, such as nutritional status, muscular mass, aerobic conditioning and emotional factors, determine their capacity for physical exertion.
No correlation was found between the general activity domain and the function tests studied. We believe that this domain is really the last to be altered in cystic fibrosis patients, and that it was due to the very good pulmonary function presented by this group of patients that no such correlation was found. The data from the physical examination correlated positively and moderately with the pulmonary function, suggesting that clinical findings that are more detailed and specific better reflect the obstructive characteristic of this disease. Nutritional status presented a significant correlation only with FEF25-75, that correlation being positive, yet moderate. In this group of patients with excellent nutritional status, the only impaired functional aspect one would expect to find would be FEF25-75, since it is the most sensitive. Correlations with FVC and FEV1 were not statistically significant, and functional loss therefore could not be correlated with nutritional status. For this aspect to affect pulmonary function, the cystic fibrosis patient would have to be in a more advanced stage of the disease. Since the radiology domain evaluates early morphological alterations of the disease, it presented positive, moderate correlations with FEF25-75 and FEV1 but not with FVC. It is known that FEV1 itself is a test with poor sensitivity for identifying mild obstructive disease. Since FVC is altered only in the later stages of the disease, it would not reflect the radiological lesions found.
The Shwachman-Kulczycki score follows the functional decline, and its correlation with spirometry data is excellent.(29) In our study, we found that the Shwachman-Kulczycki score correlates strongly with FEV1 and FEF25-75, whereas it correlates only moderately with FVC, indicating that this scoring system follows pulmonary function in its decline and that the correlation is strongest with FEF25-75, which is the most sensitive test.
The ideal radiological scoring system should reflect clinical status and functional loss.(17,26) Excellent correlations between pulmonary function and radiology have been described in the literature.(19,21,22,30) When the Brasfield score and pulmonary function were correlated, correlation coefficient values ranged from 0.50 to 0.70 for FEV1; 0.35 to 0.68 for FVC, compared with 0.74 for the correlation with FEF25-75.(12,21,22) This great variability, however, indicates that many radiological alterations cannot be detected through function tests.(12) Among the radiological scoring systems, the Brasfield score presents the highest correlation with function tests. However, probably due to the small sample size and to the high function test means, we found the Brasfield score to correlate only moderately with FVC, FEV1 and FEF25-75 (r = 0.36, r = 0.36 and r = 0.35, respectively), indicating that not all morphological alterations are reflected in pulmonary function.
The Brasfield and Shwachman-Kulczycki scores have been shown to correlate significantly with each other.(10,17,21,22,24) The strong, positive correlation found in our study (r = 0.62) demonstrates that the clinical status of the patient deteriorated in parallel with the chest X-ray findings. According to our results, there is a strong, positive correlation between the Brasfield score and the radiology domain of the Shwachman-Kulczycki system. The former is ideally performed by a pediatric radiologist; the second is performed at the medical office, by the primary care physician. In view of this strong correlation, the radiology domain can be a good, sensitive alternative in the evaluation of radiological alterations of cystic fibrosis patients when the Brasfield score is not available. This demonstrates the value of the Shwachman-Kulczycki score when measured during the medical visit, immediately providing an overall analysis of the cystic fibrosis patient.
In conclusion, the Shwachman-Kulczycki clinical score can, in isolation, reflect the clinical status of a patient, and its importance lies in the fact that it correlates positively with the Brasfield radiological score and with function tests. Although these scores assess different aspects of pulmonary involvement, their correlations make adequate monitoring possible, even in the absence of one of these methods. Our study was limited by the small sample size and by its retrospective nature. Nevertheless, our data corroborate those in the literature and contribute to expanding knowledge of patients under follow-up treatment at a cystic fibrosis referral center in Brazil.
References 1. Hubert D. Mucoviscidose. In: Encycl Méd Chir-Pneumologie. Paris: Editions Techniques; 1993. vol. 4 (6-040-L-25). p. 1-9.
2. Boucher RC. Cystic Fibrosis. In: Harrison TR, Fauci AS, editors. Harrison's principles of internal medicine. New York: McGraw-Hill; 1994. p. 1194-7.
3. Cystic Fibrosis Mutation Database. [database on the Internet]. Cystic Fibrosis Genetic Analysis Consortium [updated 2006 Sep 8; cited 2007 Feb 15]. Available from: http://www.genet.sickkids.on.ca
4. Blic J, Le Bourgeois M, Hubert D. Mucoviscidose. In: Encycl Méd Chir-Pneumologie. Paris: Editions Scientifiques et Medicales Elsevier; 2001. vol. 5 (6-040-L-25). p.1-14.
5. Robinson C, Scalin TF. Cystic Fibrosis. In: Fishman AP, Elias JA, editors. Fishman's Pulmonary Diseases and Disorders. 3rd ed. New York: McGraw-Hill, Health Professions Division; 1997. p. 803-24.
6. Abreu e Silva FA, Palombini BC. Fibrose Cística. In: Silva LC. Compêndio de pneumologia. São Paulo: Fundo Editorial Byk; 1993. p. 977-84.
7. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998;132(4):589-95.
8. I Consenso Brasileiro de Espirometria. J Pneumol. 1996;22(3):105-44.
9. Ruppel G. Manual of pulmonary function testing. 6th ed. St. Louis: Mosby; 1994. p. 43-81; 208-15.
10. Grum CM, Lynch JP 3rd. Chest radiographic findings in cystic fibrosis. Semin Respir Infect. 1992;7(3):193-209.
11. Wood RE. Prognosis. In: Taussig LM, editor. Cystic Fibrosis. New York: Thieme-Stratton; 1984. p. 434-60.
12. Sawyer SM, Carlin JB, DeCampo M, Bowes G. Critical evaluation of three chest radiograph scores in cystic fibrosis. Thorax. 1994;49(9):863-6.
13. Weatherly MR, Palmer CG, Peters ME, Green CG, Fryback D, Langhough R, et al. Wisconsin cystic fibrosis chest radiograph scoring system. Pediatrics. 1993;91(2):488-95.
14. Brouard J, Lecoq I, Viel JF, Guillot M, Laurans M, Laroche D, et al. [Evaluation of diagnosis and follow-up in screened children with cystic fibrosis in Normandy] [Article in French]. Arch Pediatr. 2001;8 Suppl 3:603-609.
15. Santos CI, Ribeiro JD, Ribeiro AF, Hessel G. Critical analysis of scoring systems used in the assessment of Cystic Fibrosis severity: State of the art. J Bras Pneumol. 2004;30(3):286-98.
16. Shwachman H, Kulczycki LL. Long-term study of one hundred five patients with cystic fibrosis; studies made over a five- to fourteen-year period. AMA J Dis Child. 1958;96(1):6-15.
17. Brasfield D, Hicks G, Soong S, Tiller RE. The chest roentgenogram in cystic fibrosis: a new scoring system. Pediatrics. 1979;63(1):24-9.
18. Assis I, Camargos PA, Reis FJ, Sulmonett N, Carneiro AP. Assessing correlations between spirometry and Shwachman-Kulczycki score in children and adolescents. Pediatr Pulmonol. 2003;36(4):305-9.
19. Matthew DJ, Warner JO, Chrispin AR, Norman AP. The relationship between chest radiographic scores and respiratory function tests in children with cystic fibrosis. Pediatr Radiol. 1977;5(4):198-200.
20. Mukhopadhyay S, Kirby ML, Duncan AW, Carswell F. Early focal abnormalities on chest radiographs and respiratory prognosis in children with cystic fibrosis. Br J Radiol. 1996;69(818):122-5.
21. Reilly BJ, Featherby EA, Weng TR, Crozier DN, Duic A, Levison H. The correlation of radiological changes with pulmonary function in cystic fibrosis. Radiology. 1971;98(2):281-5.
22. Rosenberg SM, Howatt WF, Grum CM. Spirometry and chest roentgenographic appearance in adults with cystic fibrosis. Chest. 1992;101(4):961-4.
23. Terheggen-Lagro S, Truijens N, van Poppel N, Gulmans V, van der Laag J, van der Ent C. Correlation of six different cystic fibrosis chest radiograph scoring systems with clinical parameters. Pediatr Pulmonol. 2003;35(6):441-5.
24. Wong EB, Regnis J, Shnier RC, Bye PT, Stewart ME. The relationship between tests of lung function and three chest radiological scoring systems in patients with cystic fibrosis. Australas Radiol. 1993;37(3):265-9.
25. Zapletal A. Lung function in children and adolescents. Methods, Reference Values. In: Zapletal A, Samanak M, Paul T, editors. Progress in respiration research. Basel: Karger; 1987. p. 10-45.
26. Conway SP, Pond MN, Bowler I, Smith DL, Simmonds EJ, Joanes DN, et al. The chest radiograph in cystic fibrosis: a new scoring system compared with the Chrispin-Norman and Brasfield scores. Thorax. 1994 Sep;49(9):860-2. Erratum in: Thorax 1994;49(11):1184.
27. Shwachman H, Kowalski M, Khaw KT. Cystic fibrosis: a new outlook. 70 patients above 25 years of age. Medicine (Baltimore). 1977;56(2):129-49.
28. Taussig LM, Kattwinkel J, Friedewald WT, Di Sant'Agnese PA. A new prognostic score and clinical evaluation system for cystic fibrosis. J Pediatr. 1973;82(3):380-90.
29. Camargos PA, Queiroz MV. [Peak expiratory flow rate in the management of cystic fibrosis] [Article in Portuguese]. J Pediatr (Rio J). 2002;78(1):45-9.
30. Kraemer R, Rüdeberg A, Kläy M, Rossi E. Relationship between clinical conditions, radiographic findings and pulmonary functions in patients with cystic fibrosis. Helv Paediatr Acta. 1979;34(5):417-28.
____________________________________________________________________________________________________________________
Study carried out at the Pediatric Pulmonology Department of the Porto Alegre Hospital de Clínicas, Porto Alegre, Brazil.
1. Pulmonologist and Phthisiologist. Federal University of Rio Grande do Sul, Porto Alegre, Brazil.
2. Adjunct Professor in the Pediatrics Department. Universidade Federal do Rio Grande do Sul - UFRGS, Federal University of Rio Grande do Sul - School of Medicine, Porto Alegre, Brazil.
3. Pediatric Radiologist. Conceição Children's Hospital, Porto Alegre, Brazil.
Correspondence to: Ivanice Duarte Freire. Rua Duque de Caxias, 586/401, Centro, CEP 90010-280, Porto Alegre, RS, Brasil.
Tel 55 51 3023-7018. E-mail: ivanicedf@terra.com.br
Submitted: 19 February 2007. Accepted, after review: 30 July 2007.











 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket